

Lights and Shadows on the Sourcing of Silver Radioisotopes for Targeted Imaging and Therapy of Cancer: Production Routes and Separation Methods

Abstract
:1. Introduction

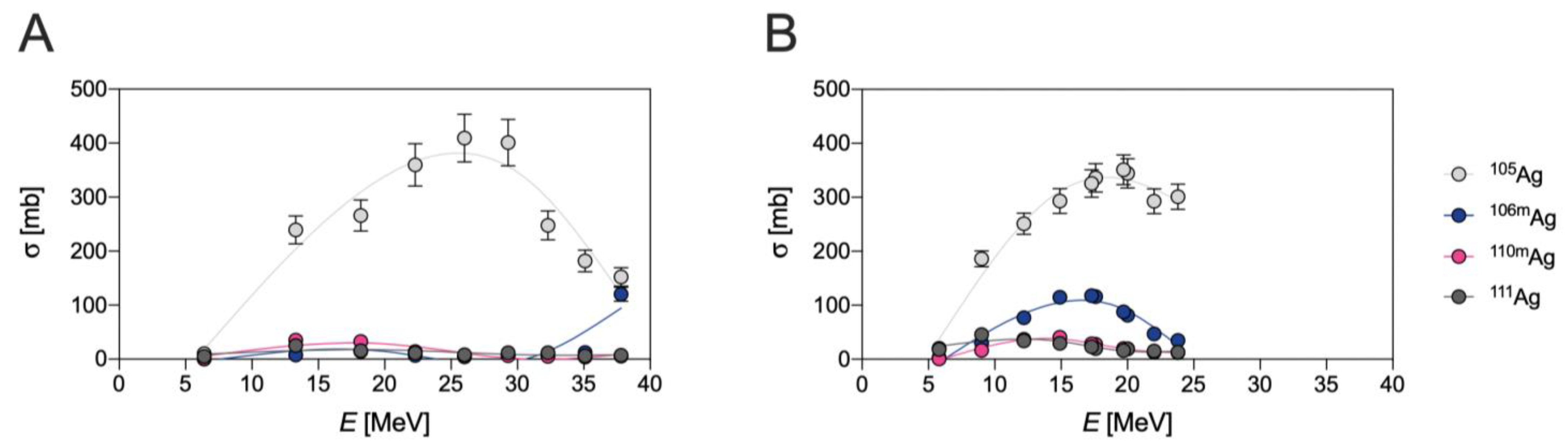{kind=link}
{kind=link}
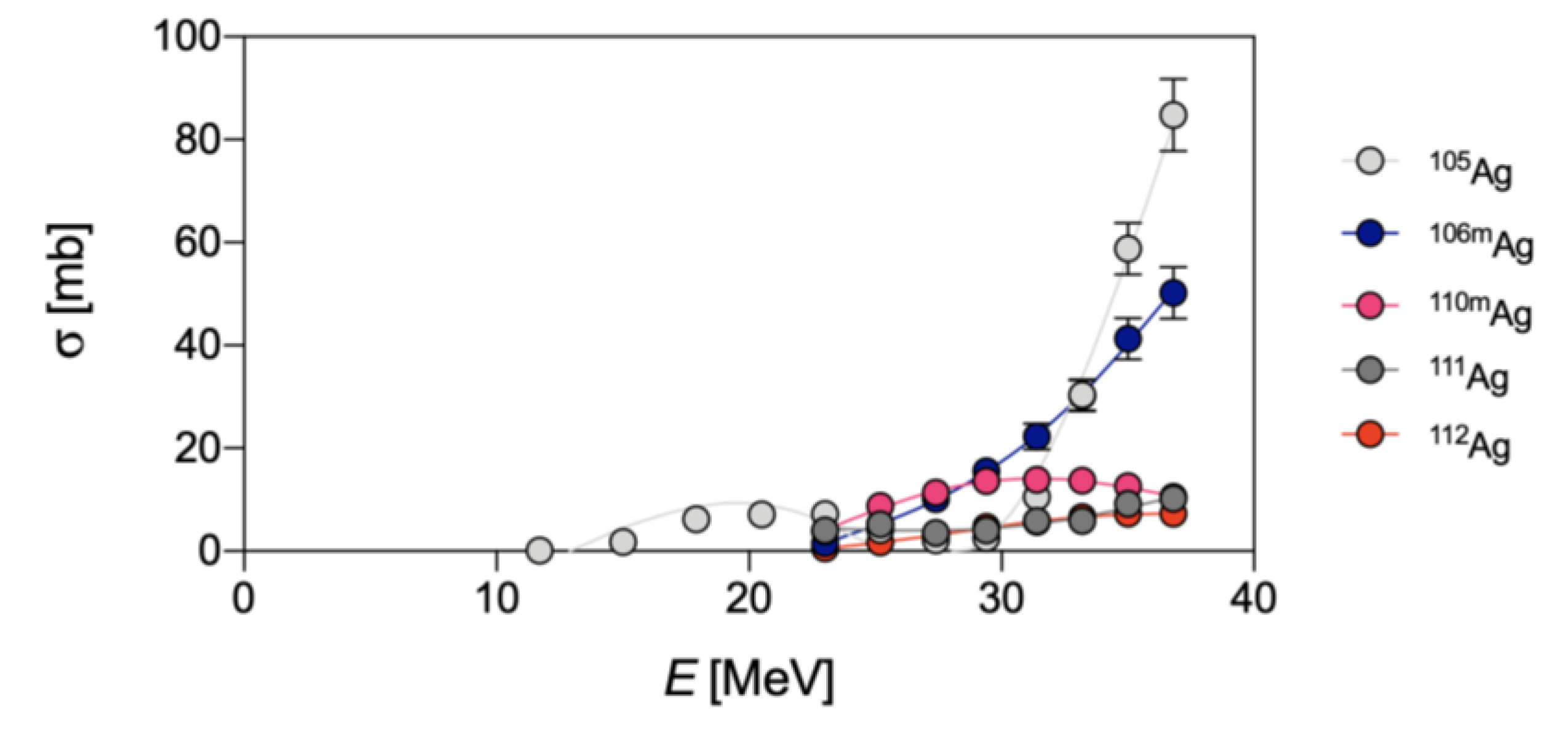{kind=link}
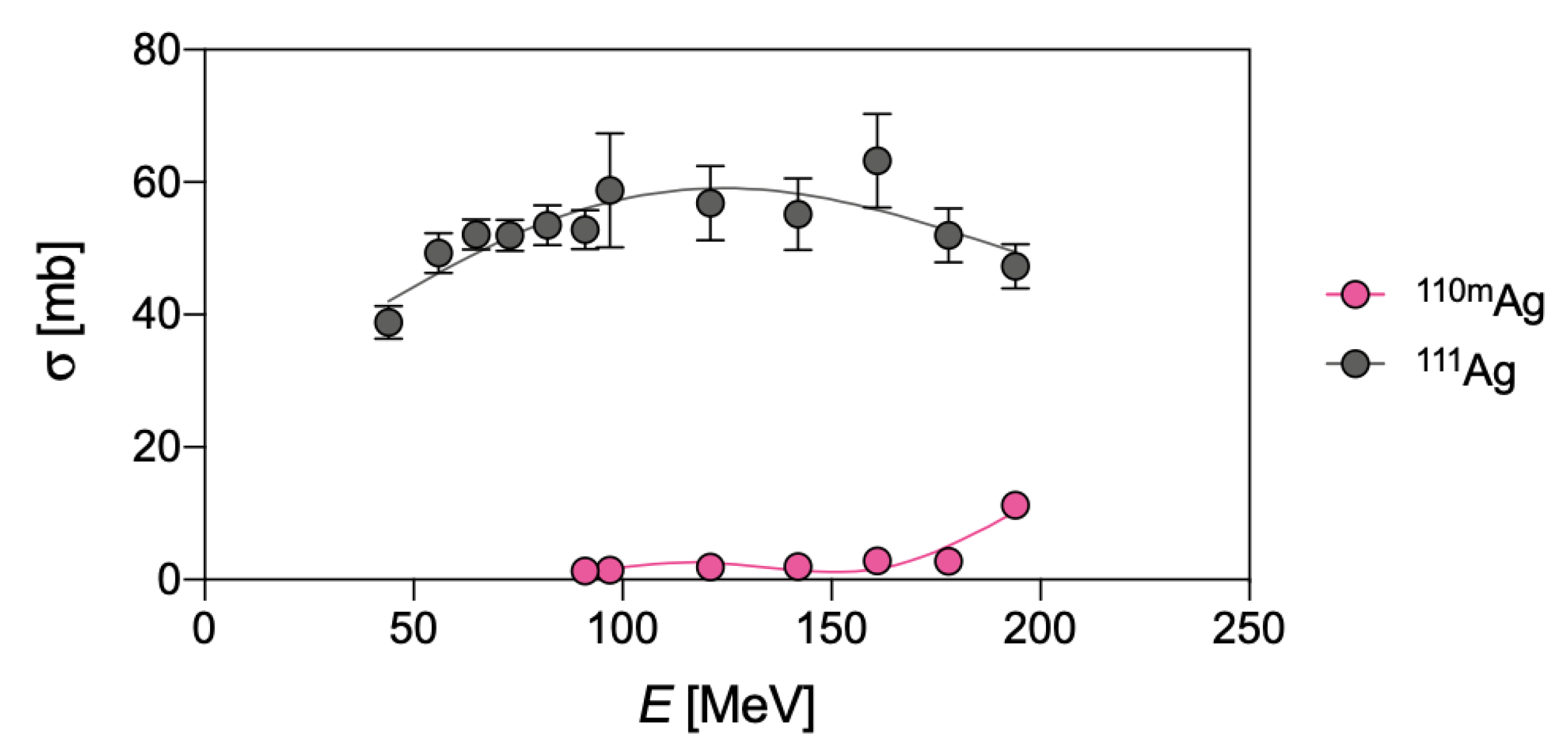{kind=link}
{kind=link}
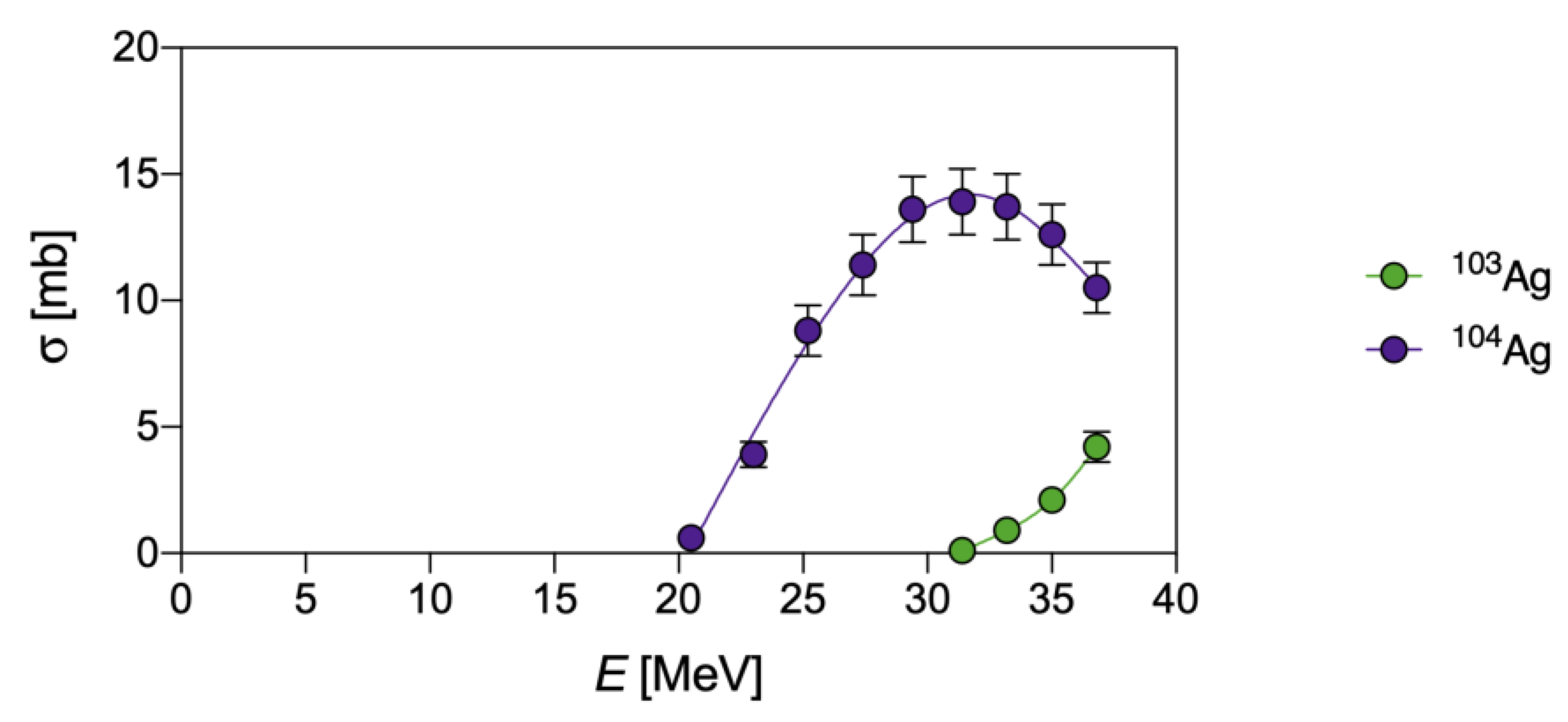{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radioisotope | Use | t1/2 | Decay Mode | Eβ− [keV] | Iβ− [%] | Eβ+ [keV] | Iβ+ [%] | Eγ [keV] | Iγ [%] |
|---|---|---|---|---|---|---|---|---|---|
| 103Ag | PET | 65.7 min | ε + β+ (100%) | - | - | 2421 | 14.8 | 118.7 | 31.2 |
| 2688 | 8 | 148.2 | 28.3 | ||||||
| 2444 | 2.1 | 266.9 | 13.3 | ||||||
| 2156 | 1.6 | 1273.8 | 9.4 | ||||||
| 104gAg | PET | 69.2 min | ε + β+ (100%) | - | - | 2014 | 5.1 | 555.8 | 92.6 |
| 2197 | 4.1 | 767.6 | 65.7 | ||||||
| 2097 | 2.9 | 941.6 | 25.0 | ||||||
| 2955 | 2.0 | 925.9 | 12.5 | ||||||
| 104mAg | PET | 33.5 min | ε + β+ + IT (100%) | 3730 | 58.0 | 555.8 | 91 | ||
| - | - | 2492 | 1.8 | 1341.7 | 1.6 | ||||
| 2944 | 1.1 | 767.6 | 0.9 | ||||||
| 111Ag | Therapy (+ SPECT) | 7.4 d | β− (100%) | 1035 | 92.0 | - | - | 342.1 | 6.7 |
| 694.7 | 7.1 | 245.4 | 1.2 | ||||||
| 791.4 | 1.0 |
2. Production of Silver-111
2.1. Reactor-Based Production
2.2. Accelerator-Based Production
2.2.1. Charged-Particle-Based Production
2.2.2. Photonuclear-Based Production
3. Production of Silver-103 and Silver-104m,g
3.1. Silver-103
3.2. Silver-104m,g
4. Separation of Silver Radioisotopes
4.1. Separation by Chemical Methods
4.1.1. Separation of Silver-111 from Palladium Targets
Chromatographic Methods
- Cation Exchange: Mansur et al. reported a cation-exchange-based chromatographic separation of 111Ag from the neutron-irradiated Pd matrix [21]. According to their protocol, the Pd target (100 mg) was dissolved in aqua regia (5 mL), followed by evaporation to dryness [21]. The process was repeated by adding HCl to remove residual amounts of HNO3 and the bulk was dissolved in distilled water (10 mL). Concentrated NH3 (25%, 7–8 mL) was then slowly added, and the resulting solution was warmed and passed through a column (1 cm diameter × 10 cm) filled with AG50W-X8 (50–100 mesh, H+ form, 5 g) pre-washed with water. A 1 mL/min flow rate was used throughout the process [21]. Silver and palladium cations were therefore adsorbed as [Ag(NH3)2]+ and [Pd(NH3)4]2+ complexes. After a washing step with water (20 mL), necessary to remove the excess of NH3, 111Ag-containing residue was eluted with a 0.5 M NaCl solution (16 mL, 80% yield) as [AgCl3]2−. Palladium was recovered by eluting the resin with 14 M HNO3 (80 mL). The concentration of Pd2+ in the 111Ag+ eluate was <1 μg/mL.
- Anion Exchange: Taylor et al. reported an anion-exchange-based separation [23]. After the irradiation (4–6 days, neutron flux = 1012 n/cm2/s), the palladium target was dissolved in aqua regia, and the resulting solution was evaporated to dryness. After heating, the residue was dissolved in a small volume of 10 M HCl to remove traces of HNO3 and passed into a column (1 cm diameter × 25 cm) filled with a Deacidite FF anion exchange resin. 111Ag-chloro complexes were eluted from the column with a subsequent rinse with 10 M HCl (50 mL). The solution was then evaporated to dryness and the residue dissolved in diluted HNO3, recovering 75% of the starting activity.
- Alumina Adsorption: Khalid et al. reported the possible use of alumina to adsorb the produced 111Ag and separate it from a neutron-irradiated palladium bulk matrix [28]. In their work, the irradiated Pd target (100 mg) was dissolved in aqua regia (5 mL) and the solution was evaporated to dryness. Repeated additions of concentrated HCl were performed to expel traces of HNO3 and the evaporation was carried out again. Then, the obtained residue was dissolved in 0.01 M HCl (30 mL) and the solution was passed at 1 mL/min through an alumina-containing column (5 g, 1 cm diameter × 10 cm) pre-conditioned with 0.01 M HCl. The column was washed with 0.1 M HCl (60 mL) to remove Pd2+ and the 111Ag-labelled residue was eluted with 4 M HCl (30–40 mL). More than 80% of the 111Ag-fraction was recovered in 20 mL and the palladium concentration was estimated to be <1 μg/mL.
Liquid/Liquid Extraction
Precipitation
- Precipitation as silver chloride: Collin et al. reported the dissolution of an irradiated palladium wire in hot concentrated HNO3 spiked with one drop of HCl [31]. Radioactive silver was then co-precipitated with an amount of stable AgNO3, inserted to increase the total mass of silver, by the addition of NaCl. Filtration allowed the recovery of AgCl, which was then dissolved in NH3 solution and reduced back to metallic silver by using ascorbic acid.

- Co-precipitation with mercury(I) chloride: Haymond et al. reported the separation of radioactive silver radioisotopes (105Ag, 106Ag and 111Ag) produced by bombarding a palladium target with 19 MeV deuterons (200 μA/h, average beam intensity 20 μA) through a precipitation technique using mercury(I) chloride as co-precipitant [16]. After the bombardment, the irradiated surface of the palladium target (approximately 0.5 g of Pd) was milled off and dissolved in aqua regia. The solution was evaporated to dryness and dissolved in 0.5 M HCl (500 mL) containing 50 mg of rhodium and ruthenium hold-back carrier. Then, a saturated solution of HgNO3 (0.5 mL) was added and the mixture was vigorously stirred. The precipitate of Hg2Cl2, containing over 95% of the radioactive silver, was centrifuged, washed with 0.5 M HCl and dissolved in the minimum needed volume of 16 M HNO3 [16]. Na2SO4 (200 mg) was then added, and the solution was evaporated to dryness (450 °C, 2 h) to drive off the mercury carrier. The residue was quantitatively solubilized in distilled water (10 mL) to give an isotonic saline solution of radioactive silver.
Co-Crystallization
Electrodeposition
Other Techniques
4.1.2. Separation of Silver-111 from Other Targets or Contaminants
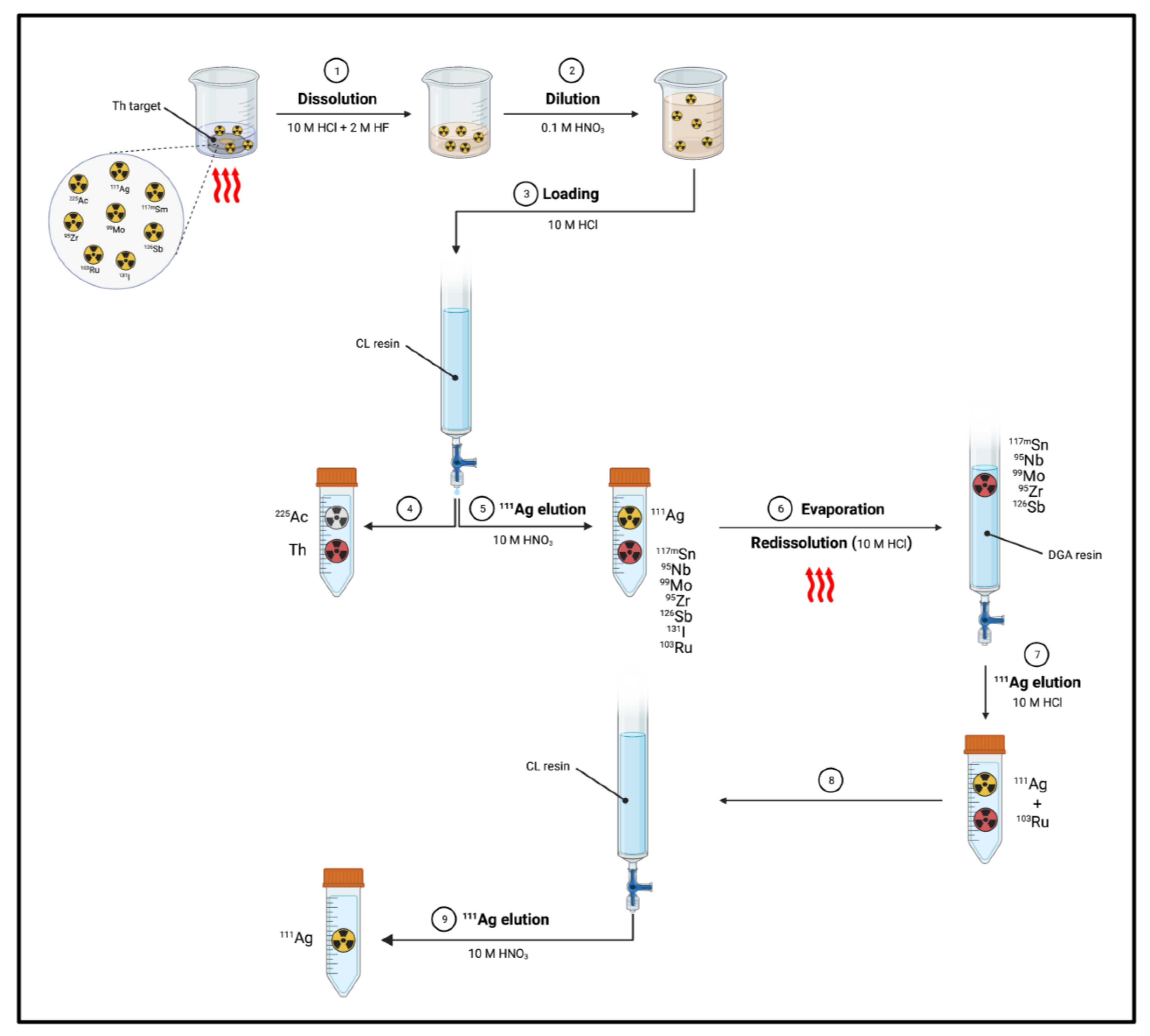
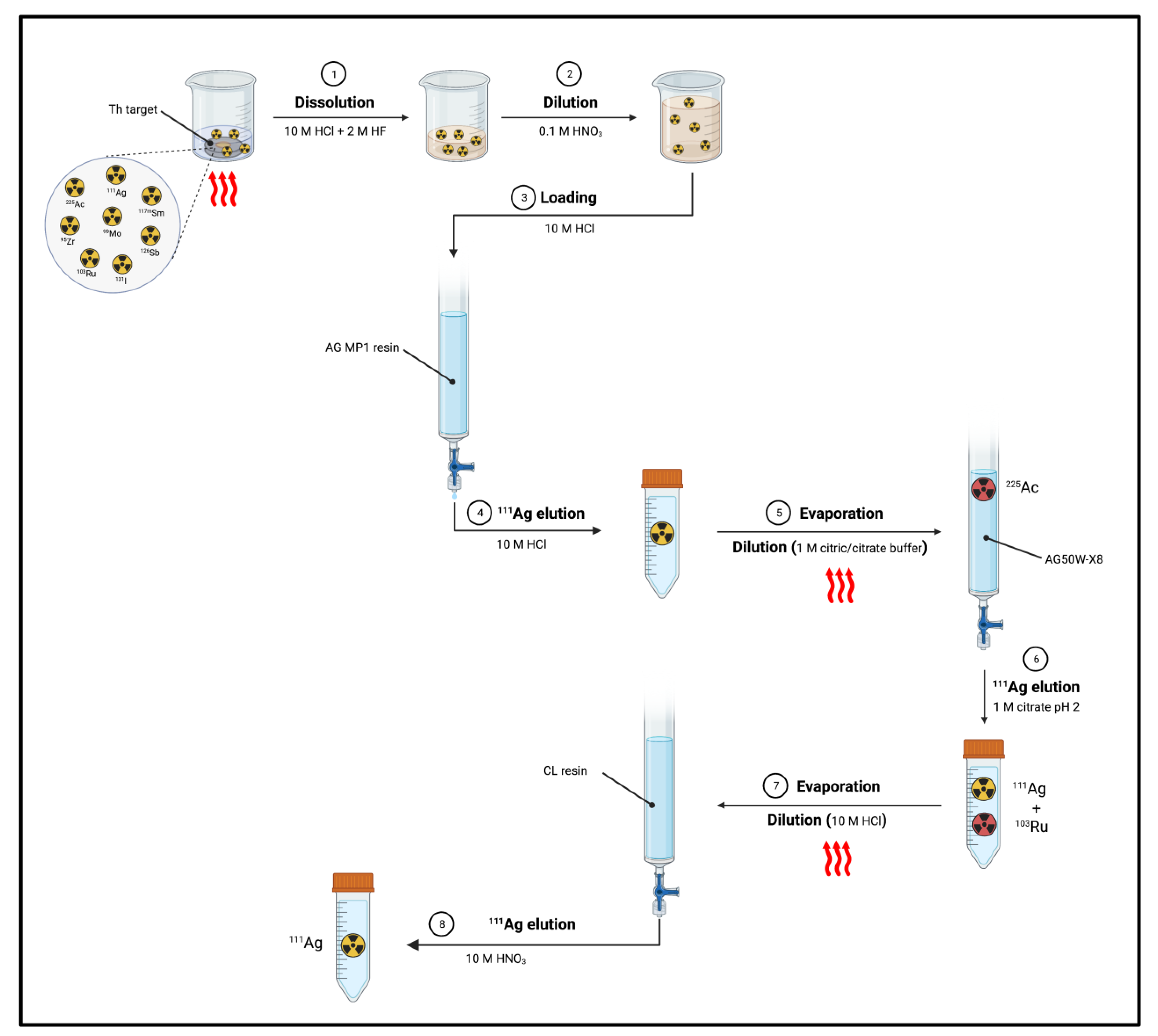
- Separation of 111Ag from Th target: Mastren et al. reported the recovery of 111Ag from a proton-irradiated thorium matrix using a solvent-impregnated CL resin composed of alkyl phosphine sulfides and alkyl phosphine oxides [1]. The author proposed two methods, both of them involving the dissolution of the irradiated thorium target (10 g) in a mixture of 10 M HCl (200 mL) and 2 M HF (0.1 mL) by heating at 80–90 °C for 2 h. Then, in the first method (Figure 11), an aliquot of the dissolved target (0.1 mL) was diluted with 0.1 M HNO3 (5 mL). Subsequently, 50 μL of the obtained solution was diluted with 10 M HCl (5 mL) and passed through a column containing the preconditioned (10 M HCl) CL resin (1 mL). 225Ac produced from irradiation and the bulk Th target were not retained by the resin, whilst 111Ag was blocked and then eluted by using 10 M HNO3 (5 mL).

- Separation of 111Ag from 111Cd: Stable 111Cd is the main isobaric contaminant remaining when 111Ag is obtained via the isotope mass separation on-line technique (ISOL—vide infra). To efficiently separate Ag+ from Cd2+ and selectively harvest 111Ag, Tosato et al. employed an extraction chromatographic resin (CL resin) [37] and developed three alternative separation methods. In the first one, upon the loading of the resin, Cd2+ was quantitatively removed in the washing step (0.1 M HNO3) whereas Ag+ was firstly retained and subsequently eluted with 10 M HNO3. The reported recovery yield was 90 ± 5% [37]. In the second separation method, 7 M NH3 was used to quantitatively elute Ag+ instead of 10 M HNO3. Finally, 1 M H2SO4 was employed in the washing step of the third method to selectively remove Cd2+, while the elution of Ag+ was conducted using 0.1 M thiourea (yield 92%). Although these methods hold promise in depleting Cd contamination from ISOL-produced 111Ag, they have yet to be tested using irradiated samples.
4.2. Mass Separation Method
5. Final Remarks and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mastren, T.; Radchenko, V.; Engle, J.W.; Weidner, J.W.; Owens, A.; Wyant, L.E.; Copping, R.; Brugh, M.; Meiring, N.; Birnbaum, E.R.; et al. Chromatographic Separation of the Theranostic Radionuclide 111Ag from a Proton Irradiated Thorium Matrix. Anal. Chim. Acta 2018, 998, 75–82. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, J.A.; Bardiès, M.; Wheldon, T.E. Relationships between Tumor Size and Curability for Uniformly Targeted Therapy with Beta-Emitting Radionuclides. J. Nucl. Med. 1995, 36, 1902–1909. [Google Scholar] [PubMed]
- Hermanne, A.; Takacs, S.; Tarkanyi, F.; Bolbos, R. Experimental Cross Sections for Charged Particle Production of the Therapeutic Radionuclide 111Ag and its PET Imaging Analogue 104m,gAg. Nucl. Instrum. Methods Phys. Res. Sect. B 2004, 217, 193–201. [Google Scholar] [CrossRef]
- Ukon, N.; Aikawac, M.; Komorid, Y.; Habad, H. Production Cross Sections of Deuteron-Induced Reactions on Natural Palladium for Ag Isotopes. Nucl. Instrum. Methods Phys. Res. Sect. B 2018, 426, 13–17. [Google Scholar] [CrossRef]
- Gyr, T.; Mäcke, H.R.; Hennig, M. A Highly Stable Silver(I) Complex of a Macrocycle Derived from Tetraazatetrathiacyclen. Angew. Chem. 1997, 36, 2786–2788. [Google Scholar] [CrossRef]
- Tosato, M.; Asti, M.; Dalla Tiezza, M.; Orian, L.; Häussinger, D.; Vogel, R.; Köster, U.; Jensen, M.; Andrighetto, A.; Pastore, P.; et al. Highly Stable Silver(I) Complexes with Cyclen-Based Ligands Bearing Sulfide Arms: A Step Toward Silver-111 Labeled Radiopharmaceuticals. Inorg. Chem. 2020, 59, 10907–10919. [Google Scholar] [CrossRef]
- Tosato, M.; Asti, M.; Di Marco, V.; Jensen, M.L.; Schell, J.; Dang, T.T.; Köster, U.; Jensen, M.; Hemmingsen, L. Towards In Vivo Applications of 111Ag Perturbed Angular Correlation of γ-Rays (PAC) Spectroscopy. Appl. Rad. Isot. 2022, 190, 110508. [Google Scholar] [CrossRef]
- Blackadar, C.; Choi, K.Y.G.; Embree, M.F.; Hennkens, H.M.; Rodríguez-Rodríguez, C.; Hancock, R.E.W.; Saatchi, K.; Häfeli, U.O. SPECT/CT Imaging of 111Ag for the Preclinical Evaluation of Silver-Based Antimicrobial Nanomedicines. ACS Appl. Mater. Interfaces 2022, 14, 26382–26393. [Google Scholar] [CrossRef]
- Aweda, T.A.; Zhang, S.; Mupanomunda, C.; Burkemper, J.; Heo, G.S.; Bandara, N.; Lin, M.; Cutler, C.S.; Cannon, C.L.; Youngs, W.J.; et al. Investigating the Pharmacokinetics and Biological Distribution of Silver-Loaded Polyphosphoester-Based Nanoparticles Using 111Ag as a Radiotracer. J. Label. Comp. Radiopharm. 2015, 58, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Aweda, T.A.; Ikotun, O.; Mastren, T.; Cannon, C.L.; Wright, B.; Youngs, W.J.; Cutler, C.; Guthrie, J.; Lapi, S.E. The Use of 111Ag as a Tool for Studying Biological Distribution of Silver-Based Antimicrobials. Medchemcomm 2013, 4, 1015–1017. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Vimalnath, K.V.; Saha, S.; Korde, A.; Sarma, H.D.; Pal, S.; Das, M.K. Preparation and Evaluation of a New Radiopharmaceutical for Radio Synovectomy, Ag-111-Labelled Hydroxyapatite (HA) Particles. Appl. Radiat. Isot. 2008, 66, 334–339. [Google Scholar] [CrossRef]
- Available online: www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html (accessed on 20 November 2022).
- Alberto, R.; Blauenstein, P.; Novak-Hofer, I.; Smith, A.; Schubiger, P.A. An Improved Method for the Separation of Ag-111 from Irradiated Natural Palladium. Appl. Radiat. Isot. 1992, 43, 869–872. [Google Scholar] [CrossRef]
- El-Azony, K.M.; Mohamed, N.M.A.; Aloraini, D.A. Advantages and Disadvantages of Nuclear Reactions Used in Reactors or Cyclotrons, in Addition to a Theoretical Study Based on Photodisintegration on Natural Indium for 111Ag Production. Nucl. Sci. Tech. 2022, 33, 14. [Google Scholar] [CrossRef]
- Morselli, L.; Donzella, A.; Arzenton, A.; Asti, M.; Bortolussi, S.; Corradetti, S.; D’Agostino, G.; Luzio, M.D.; Ferrari, M.; Gandini, A.; et al. Production and Characterization of 111Ag Radioisotope for Medical Use in a TRIGA Mark II Nuclear Research Reactor. Appl. Radiat. Isot. 2023, 197, 110798. [Google Scholar] [CrossRef]
- Haymond, H.R.; Larson, K.H.; Maxwell, R.D.; Garrison, W.M.; Hamilton, J.G. Carrier-Free Radioisotopes from Cyclotron Targets. VI. Preparation and Isolation of Ag105,106,111 from Palladium. J. Chem. Phys. 1950, 18, 391–392. [Google Scholar] [CrossRef]
- Ditrói, F.; Tárkányi, F.; Takács, S.; Hermanne, A.; Ignatyuk, A.V.; Baba, M. Activation Cross-Sections of Deuteron Induced Reactions on Natural Palladium. Nucl. Instrum. Methods Phys. Res. Sect. B 2012, 270, 61–74. [Google Scholar] [CrossRef]
- Hermanne, A.; Tárkányi, F.; Takács, S.; Shubin, Y.N. Experimental Determination of Cross Section of Alpha-Induced Reactions on natPd. Nucl. Instrum. Methods Phys. Res. Sect. B 2005, 229, 321–332. [Google Scholar] [CrossRef]
- Hermanne, A.; Takács, S.; Tárkányi, F.; Bolbos, R. Cross Section Measurements of Proton and Deuteron Induced Formation of 103Ag in Natural Palladium. Radiochim. Acta 2004, 92, 215–218. [Google Scholar] [CrossRef]
- Tárkányi, F.; Ditrói, F.; Takács, S.; Csikai, J.; Hermanne, A.; Uddin, M.S.; Baba, M. Activation Cross Sections of Proton Induced Nuclear Reactions on Palladium up to 80 MeV. Appl. Radiat. Isot. 2016, 114, 128–144. [Google Scholar] [CrossRef] [Green Version]
- Mansur, M.S.; Mushtaq, A.; Muhammad, A. Separation of 111Ag from Neutron Irradiated Natural Palladium. Radiochim. Acta 1995, 68, 161–162. [Google Scholar] [CrossRef]
- Lyle, S.J.; Maghzian, R. Separation of Carrier-Free Silver from Neutron-Irradiated Palladium. Talanta 1968, 15, 712–713. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M. The Preparation of Carrier-Free Silver-111. Int. J. Appl. Radiat. Isot. 1961, 12, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Vimalnath, K.V.; Chirayil, V.; Saha, S. Studies on the Preparation of 109Pd and 111Ag by (n,γ) Reactions on Natural Palladium for Possible Applications in Radionuclide Therapy. Proc. DAE-BRNS Symp. Nucl. Radiochem. 2007, 39, 11. [Google Scholar]
- Ohya, T.; Nagatsu, K.; Hanyu, M.; Minegishi, K.; Zhang, M.R. Separation of Radiosilver from a Cyclotron-Irradiated Palladium Target. Radiochim. Acta 2020, 108, 641–648. [Google Scholar] [CrossRef]
- Bauer, R.; Danielsen, E.; Hemmingsen, L.; Bjerrum, M.J.; Hansson, Ö.; Singh, K. Interplay between Oxidation State and Coordination Geometry of Metal Ions in Azurin. J. Am. Chem. Soc. 1997, 119, 157–162. [Google Scholar] [CrossRef]
- Ooe, K.; Watabe, T.; Shirakami, Y.; Mori, D.; Yokokita, T.; Komori, Y.; Haba, H.; Hatazawa, J. Chemical Separation of Theranostic Radionuclide 111Ag Produced in natPd(d,x)111Ag Reactions. RIKEN Accel. Prog. Rep. 2019, 53, 196. [Google Scholar]
- Khalid, M.; Mushtaq, A.; Iqbal, M.Z. Separation of 111Ag from Neutron Irradiated Natural Palladium Using Alumina as an Adsorbent. Appl. Radiat. Isot. 2000, 52, 19–22. [Google Scholar] [CrossRef]
- Lahiri, S.; Mukhopadhyay, B.; Nandy, M.; Das, N.R. Sequential Separation by HDEHP of Carrier-Free 101,105,106Rh, 103,104,105,106,110,112Ag and 104,105,107,109,111Cd Produced in Alpha-Particle Activated Palladium. J. Radioanal. Nucl. Chem. 1997, 224, 155–158. [Google Scholar] [CrossRef]
- Lahiri, S.; Nandy, M.; Mukhopadhyay, B. Sequential Separation of Carrier Free Radioisotopes of Rhodium, Silver and Cadmium Produced in α-Particle Activated Palladium by TOA. Appl. Radiat. Isot. 1997, 48, 1169–1172. [Google Scholar] [CrossRef]
- Collins, S.; Keightley, J.; Gilligan, C.; Gasparro, J.; Pearce, A. Determination of the Gamma Emission Intensities of 111Ag. Appl. Radiat. Isot. 2014, 87, 107–111. [Google Scholar] [CrossRef]
- Sicilio, F.; Peterson, M.D.; Rudolph, G.G. Separation of Radioactive Silver-111 from Pile-Irradiated Palladium. Anal. Chem. 1956, 28, 365–366. [Google Scholar] [CrossRef]
- Zimen, K.E. Gewinnung von Radiosilber (Ag111). Z. Nat. A 1949, 4, 95–96. [Google Scholar] [CrossRef]
- Micheev, N.B.; Abdel-Rassoul, A.A.; Fouad, H. Application of Co-Crystallisation Technique for the Production of Carrier Free Silver-111 from Pile Irradiated Palladium. Z. Anorg. Allg. Chem. 1964, 332, 209–215. [Google Scholar] [CrossRef]
- Griess, J.; Rogers, L.B. Selective Electrodeposition of Silver. U.S. Patent US-2612470-A, 30 September 1952. [Google Scholar]
- Miller, J.; Toth, G. Removal of Silver Traces from Palladium by Means of Selective Adsorption. Isot. Environ. Health Stud. 1967, 3, 19–20. [Google Scholar] [CrossRef]
- Tosato, M.; Nardella, S.; Badocco, D.; Pastore, P.; Andrighetto, A.; Realdon, N.; Di Marco, V. Chemical Purification of 111Ag from Isobaric Impurity 111Cd by Solid Phase Extraction Chromatography: A Proof of Concept Study. Appl. Radiat. Isot. 2020, 164, 109263. [Google Scholar] [CrossRef]
- Ballan, M.; Vettorato, E.; Morselli, L.; Tosato, M.; Nardella, S.; Borgna, F.; Corradetti, S.; Monetti, A.; Lunardon, M.; Zenoni, A.; et al. Development of Implantation Substrates for the Collection of Radionuclides of Medical Interest Produced via ISOL Technique at INFN-LNL. Appl. Radiat. Isot. 2021, 175, 109795. [Google Scholar] [CrossRef]
- Van Duppen, P. Isotope Separation on Line and Post Acceleration. The Euroschool Lectures on Physics with Exotic Beams; Springer: Berlin/Heidelberg, Germany, 2006; Volume II, pp. 37–77. [Google Scholar]
- Andrighetto, A.; Borgna, F.; Ballan, M.; Corradetti, S.; Vettorato, E.; Monetti, A.; Rossignoli, M.; Manzolaro, M.; Scarpa, D.; Prete, G.; et al. The ISOLPHARM Project: A New ISOL Production Method of High Specific Activity Beta-Emitting Radionuclides as Radiopharmaceutical Precursors. Int. J. Mod. Phys. Conf. Ser. 2018, 48, 1860103. [Google Scholar] [CrossRef] [Green Version]
- Ballan, M.; Tosato, M.; Verona, M.; Caeran, M.; Borgna, F.; Vettorato, E.; Corradetti, S.; Zangrando, L.; Sgaravatto, M.; Verlato, M.; et al. Preliminary Evaluation of the Production of Non-Carrier Added 111Ag as Core of a Therapeutic Radiopharmaceutical in the Framework of ISOLPHARM_Ag Experiment. Appl. Radiat. Isot. 2020, 164, 109258. [Google Scholar] [CrossRef]
- Talip, Z.; Favaretto, C.; Geistlich, S.; van der Meulen, N.P. A Step-by-Step Guide for the Novel Radiometal Production for Medical Applications: Case Studies with 68Ga, 44Sc, 177Lu and 161Tb. Molecules 2020, 25, 966. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos Augusto, R.M.; Buehler, L.; Lawson, Z.; Marzari, S.; Stachura, M.; Stora, T.; CERN-MEDICIS Collaboration. CERN-MEDICIS (Medical Isotopes Collected from ISOLDE): A New Facility. Appl. Sci. 2014, 4, 265–281. [Google Scholar] [CrossRef] [Green Version]










| Isotope | Natural Abundance [%] | Nuclear Reaction and Final Product | Cross-Section [mb] |
|---|---|---|---|
| Palladium-102 | 1.02 |  | 180 |
| Palladium-104 | 11.14 |  | 75 |
| Palladium-105 | 22.33 |  | 217 |
| Palladium-106 | 27.33 |  | 29 |
| Palladium-108 | 26.46 |  | 868 |
| Palladium-110 | 11.72 |  | 340 |
| Target Material | Produced Isotope | Threshold Energy [MeV] |
|---|---|---|
| Natural Cd | 111Ag | 9.6 |
| 105Ag | 7.3 | |
| 107Ag | 8.1 | |
| 109Ag | 8.9 | |
| 110mAg | 9.1 | |
| 112Ag | 9.7 | |
| 113Ag | 10.2 | |
| 115Ag | 11.0 | |
| 107Cd | 10.3 | |
| 109Cd | 9.9 | |
| Natural In | 111Ag | 3.7 |
| 109Ag | 3.1 | |
| 114mIn | 9.0 | |
| 114Cd | 6.8 | |
| 112mIn | 9.4 | |
| 112Cd | 6.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tosato, M.; Asti, M. Lights and Shadows on the Sourcing of Silver Radioisotopes for Targeted Imaging and Therapy of Cancer: Production Routes and Separation Methods. Pharmaceuticals 2023, 16, 929. https://doi.org/10.3390/ph16070929
Tosato M, Asti M. Lights and Shadows on the Sourcing of Silver Radioisotopes for Targeted Imaging and Therapy of Cancer: Production Routes and Separation Methods. Pharmaceuticals. 2023; 16(7):929. https://doi.org/10.3390/ph16070929
Chicago/Turabian StyleTosato, Marianna, and Mattia Asti. 2023. "Lights and Shadows on the Sourcing of Silver Radioisotopes for Targeted Imaging and Therapy of Cancer: Production Routes and Separation Methods" Pharmaceuticals 16, no. 7: 929. https://doi.org/10.3390/ph16070929