
A Novel Acetylation-Immune Subtyping for the Identification of a BET Inhibitor-Sensitive Subgroup in Melanoma
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Analysis of Melanoma Cell Lines
2.2. Analysis of TCGA SKCM Datasets
2.3. Statistical Analysis
3. Results
3.1. Identification of Two ALISs of Melanoma Cell Lines
3.2. Identification of Three ALISs of Skin Cutaneous Melanoma (SKCM) Patients with Primary Tumors
3.3. Clinical Characteristics and Molecular Characteristics in Different SKCM ALISs
3.4. Immunological Characteristics across Three SKCM ALISs
3.5. Protein Interaction (PI) Network Reveals the Vital Role of KAT2A in Regulating BETi Resistance-Related Biological Genes and Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA A Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The growing burden of invasive melanoma: Projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.j.; Thompson, J.F. Update on the melanoma staging system: The importance of sentinel node staging and primary tumor mitotic rate. J. Surg. Oncol. 2011, 104, 379–385. [Google Scholar] [CrossRef]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T. Histone variant H2A. Z. 2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Tiffen, J.C.; Gowrishankar, K.; Jin, L.; Pupo, G.M.; Cullinane, C.; Prinjha, R.K.; Smithers, N. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J. Investig. Dermatol. 2014, 134, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; González-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W. BRD4 Sustains Melanoma Proliferation and Represents a New Target for Epigenetic TherapyBRD4 Is a New Therapeutic Target in Melanoma. Cancer Res. 2013, 73, 6264–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autin, P.; Blanquart, C.; Fradin, D. Epigenetic drugs for cancer and microRNAs: A focus on histone deacetylase inhibitors. Cancers 2019, 11, 1530. [Google Scholar] [CrossRef] [Green Version]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted DrugsOTX015 in Preclinical Models of Lymphomas. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Chung, C.-W.; Coste, H.; White, J.H.; Mirguet, O.; Wilde, J.; Gosmini, R.L.; Delves, C.; Magny, S.M.; Woodward, R.; Hughes, S.A. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [Google Scholar] [CrossRef]
- Erkes, D.A.; Field, C.O.; Capparelli, C.; Tiago, M.; Purwin, T.J.; Chervoneva, I.; Berger, A.C.; Hartsough, E.J.; Villanueva, J.; Aplin, A.E. The next-generation BET inhibitor, PLX51107, delays melanoma growth in a CD8-mediated manner. Pigment Cell Melanoma Res. 2019, 32, 687–696. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clément, C.A.; Barnathan, M.; Brusq, J.-M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O. Discovery of epigenetic regulator I-BET762: Lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.-P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical Response of Carcinomas Harboring the BRD4–NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628OTX015/MK-8628 Activity in Patients with NMC. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Mustafi, S.; Camarena, V.; Volmar, C.-H.; Huff, T.C.; Sant, D.W.; Brothers, S.P.; Liu, Z.-J.; Wahlestedt, C.; Wang, G. Vitamin C Sensitizes Melanoma to BET InhibitorsVitamin C Improves the Response of Melanoma to BETi. Cancer Res. 2018, 78, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Makowski, A.M.; Dutnall, R.N.; Annunziato, A.T. Effects of acetylation of histone H4 at lysines 8 and 16 on activity of the Hat1 histone acetyltransferase. J. Biol. Chem. 2001, 276, 43499–43502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, P.; Zhao, J.; Meng, Z.; Wu, H.; Wang, B.; Wu, H.; Jin, X. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Alistar, A.; Chou, J.W.; Nagalla, S.; Black, M.A.; D’Agostino, R.; Miller, L.D. Dual roles for immune metagenes in breast cancer prognosis and therapy prediction. Genome Med. 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Huang, R.S.; Duan, S.; Bleibel, W.K.; Kistner, E.O.; Zhang, W.; Clark, T.A.; Chen, T.X.; Schweitzer, A.C.; Blume, J.E.; Cox, N.J. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 9758–9763. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.-B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Joseph, E.W.; Pratilas, C.A.; Poulikakos, P.I.; Tadi, M.; Wang, W.; Taylor, B.S.; Halilovic, E.; Persaud, Y.; Xing, F.; Viale, A. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. USA 2010, 107, 14903–14908. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, D.; Almouzni, G.; Soria, J.-C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017, 28, 254–269. [Google Scholar] [CrossRef]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims III, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic StrategiesABBV-075 Induces Apoptosis and Exhibits Synergy with ABT-199. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef] [Green Version]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and in vitro evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK–ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2017, 23, 2781–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, W.; Pang, Y.; Li, R.; Wei, X.; Jiao, X.; Shi, J.; Yu, J.; Mao, H.; Liu, P. Akt/mTOR-mediated autophagy confers resistance to BET inhibitor JQ1 in ovarian cancer. OncoTargets Ther. 2019, 12, 8063. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Arede, L.; Pina, C. Buffering noise: KAT2A modular contributions to stabilization of transcription and cell identity in cancer and development. Exp. Hematol. 2021, 93, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims III, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef]
- Guo, L.; Lee, Y.-T.; Zhou, Y.; Huang, Y. Targeting epigenetic regulatory machinery to overcome cancer therapy resistance. Semin. Cancer Biol. 2022, 83, 487–502. [Google Scholar] [CrossRef]
- Li, Y.; Jaramillo-Lambert, A.N.; Yang, Y.; Williams, R.; Lee, N.H.; Zhu, W. And-1 is required for the stability of histone acetyltransferase Gcn5. Oncogene 2012, 31, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Miao, B.-P.; Zhang, R.-S.; Yang, G.; Sun, J.-J.; Tang, Y.-Y.; Liang, W.-F.; Liu, T.; Wen, Z.; Yang, P.-C.; Nie, G.-H. Histone acetyltransferase 1 up regulates Bcl2L12 expression in nasopharyngeal cancer cells. Arch. Biochem. Biophys. 2018, 646, 72–79. [Google Scholar] [CrossRef]
- Han, N.; Shi, L.; Guo, Q.; Sun, W.; Yu, Y.; Yang, L.; Zhang, X.; Zhang, M. HAT1 induces lung cancer cell apoptosis via up regulating Fas. Oncotarget 2017, 8, 89970–89977. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics a | ALIS I (N = 33) | ALIS II (N = 39) | ALIS III (N = 32) | p Value |
|---|---|---|---|---|
| Sex (N, %) | 0.659 | |||
| Male | 17 (58.6) | 20 (64.5) | 12 (52.2) | |
| Female | 12 (41.4) | 11 (35.5) | 11 (47.8) | |
| total | 29 | 31 | 23 | |
| Age () | 63.52 ± 15.05 | 62.68 ± 14.63 | 63.91 ± 13.31 | 0.939 |
| AJCC Stage (N, %) b | 0.047 | |||
| Stage I | 0 (0) | 3 (8.1) | 0 (0) | |
| Stage II | 23 (69.7) | 26 (70.3) | 18 (60) | |
| Stage III | 7 (21.2) | 8 (21.6) | 12 (40) | |
| Stage IV | 3 (9.1) | 0 (0) | 0 (0) | |
| total | 33 | 37 | 30 | |
| Neoplasm Cancer Status (N, %) | 0.020 | |||
| Tumor-free | 17 (51.5) | 30 (76.9) | 28 (87.5) | |
| With tumor | 16 (48.5) | 9 (23.1) | 4 (12.5) | |
| total | 33 | 39 | 32 | |
| Race (N, %) | 0.888 | |||
| Asian | 3 (9.1) | 2 (5.3) | 2 (6.5) | |
| White | 30 (90.9) | 36 (94.7) | 29 (93.5) | |
| total | 33 | 38 | 31 | |
| BRAF mutation (N, %) | 0.558 | |||
| Wild | 12 (42.9) | 9 (34.6) | 11 (50) | |
| Mutated | 16 (57.1) | 17 (65.4) | 11 (50) | |
| total | 28 | 26 | 22 | |
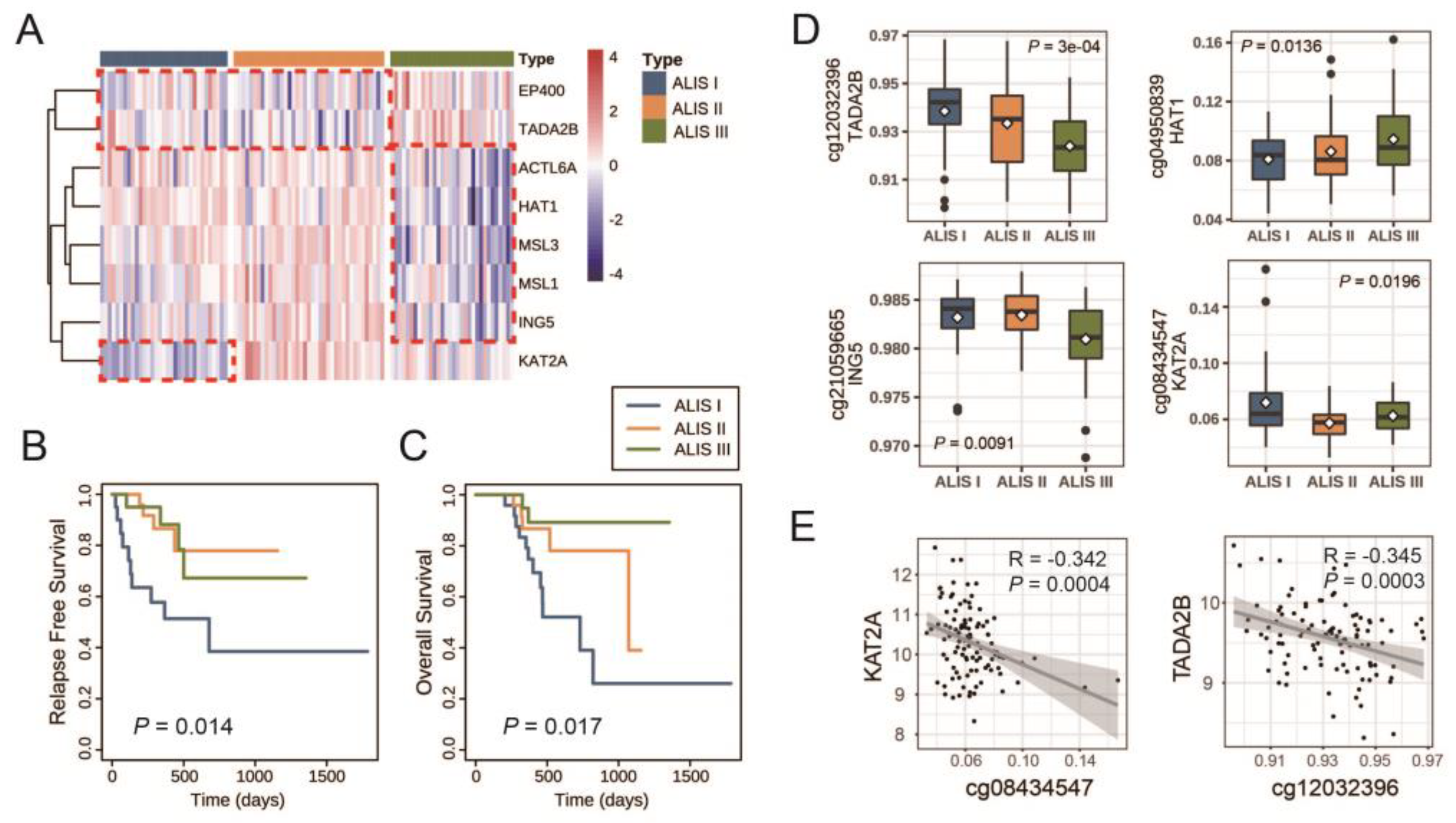
| CG Sites | Gene Symbol | p Value | I vs. II b | I vs. III b | II vs. III b |
|---|---|---|---|---|---|
| cg12032396 | TADA2B | 3 × 10−4 | 0.5050 | 0.0008 | 0.0358 |
| cg04950839 | HAT1 | 0.0136 | 0.9110 | 0.0420 | 0.3520 |
| cg21059665 | ING5 | 0.0091 | 1.0000 | 0.0210 | 0.0250 |
| cg08434547 | KAT2A | 0.0196 | 0.0160 | 0.8280 | 0.3520 |
| cg24545687 | EP400 | 0.0351 | 1.0000 | 0.1100 | 0.4300 |
| cg25198545 | ACTL6A | 0.8958 | - | - | - |
| cg09665160 | MSL1 | 0.9840 | - | - | - |
| cg20205061 | MSL3 | 0.2193 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, L.; Li, S.; Cao, L.; Li, K.; Zhao, W. A Novel Acetylation-Immune Subtyping for the Identification of a BET Inhibitor-Sensitive Subgroup in Melanoma. Pharmaceuticals 2023, 16, 1037. https://doi.org/10.3390/ph16071037
Wang L, Zhang L, Li S, Cao L, Li K, Zhao W. A Novel Acetylation-Immune Subtyping for the Identification of a BET Inhibitor-Sensitive Subgroup in Melanoma. Pharmaceuticals. 2023; 16(7):1037. https://doi.org/10.3390/ph16071037
Chicago/Turabian StyleWang, Liuying, Liuchao Zhang, Shuang Li, Lei Cao, Kang Li, and Weiwei Zhao. 2023. "A Novel Acetylation-Immune Subtyping for the Identification of a BET Inhibitor-Sensitive Subgroup in Melanoma" Pharmaceuticals 16, no. 7: 1037. https://doi.org/10.3390/ph16071037