
Agnostic Approvals in Oncology: Getting the Right Drug to the Right Patient with the Right Genomics
 , , ,
, , ,
Abstract
:1. Introduction
1.1. Brief History of a Therapeutic Paradigm Change: The Revolution in Oncology
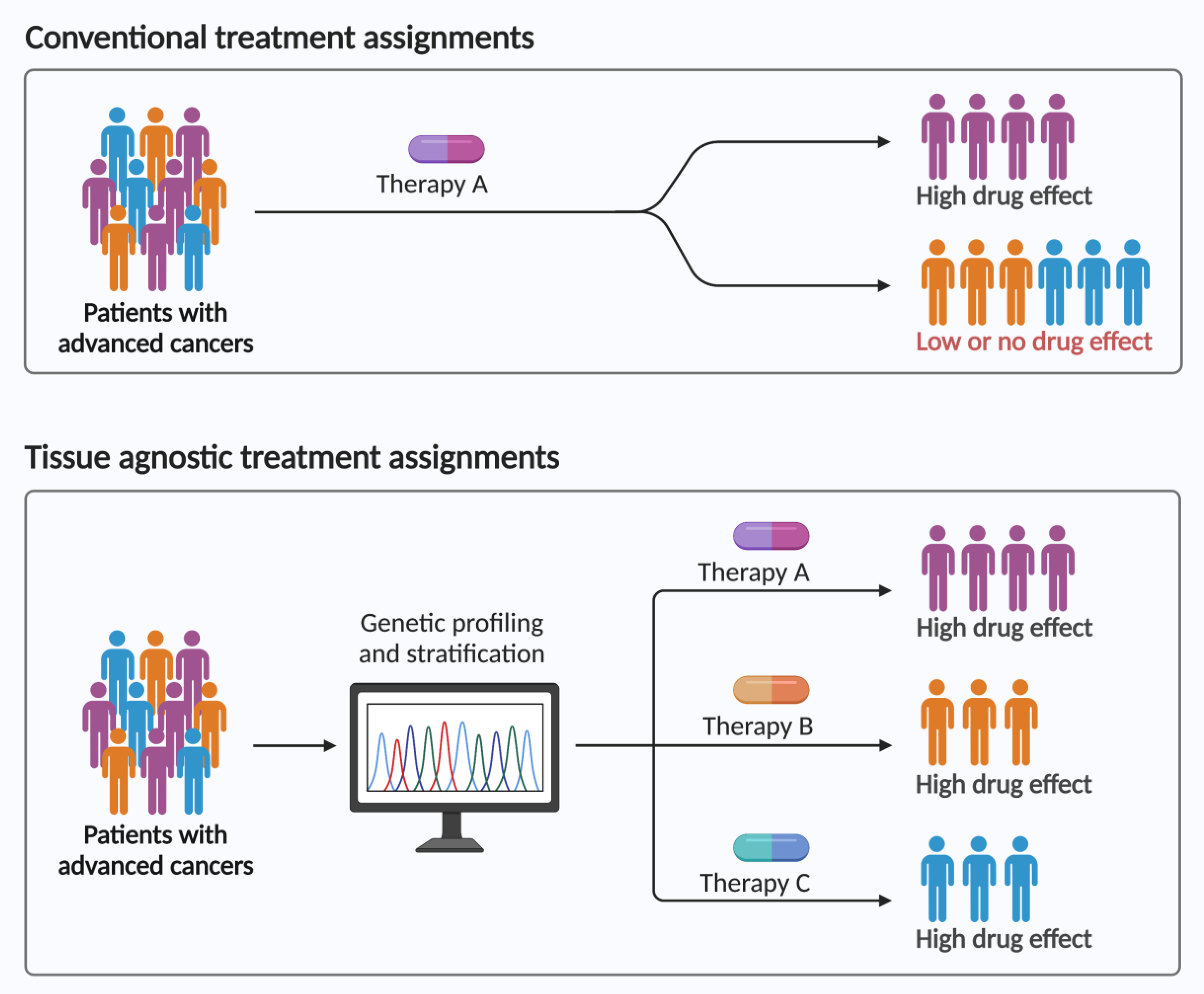
1.2. Agnostic Biomarkers and Therapies Co-Development
2. Approved Tumor-Agnostic Treatments
- 2017: pembrolizumab for patients with tumors deficient in mismatch repair (MMR) or with high microsatellite instability (MSI) (Section 2.1);
- 2018: larotrectinib for patients with neurotrophic receptor tyrosine kinase (NTRK) fusions-positive tumors (Section 2.3);
- 2019: entrectinib in patients with NTRK fusions-positive tumors (Section 2.3);
- 2020: pembrolizumab for patients affected by tumors with high tumor mutational burden (TMB) (Section 2.2);
- 2021: dostarlimab-gxly for patients with mismatch repair deficient tumors (Section 2.1);
- 2022: dabrafenib + trametinib in patients with BRAF V600E mutated tumors (Section 2.4);
- 2022: selpercatinib in patients with REarranged during Transfection (RET) fusion-positive tumors (Section 2.5).
2.1. MMRd/MSI-H: Pembrolizumab and Dostarlimab
2.2. TMB-H: Pembrolizumab
2.3. NTRK-Fusions: Larotrectinib and Entrectinib
2.4. BRAF V600E: Dabrafenib plus Trametinib
2.5. RET Fusions: Selpercatinib

{kind=link}
{kind=link}
| Drug | Target | Date of Agnostic Approval | Indication | Evidence for Approval | Common Adverse Effects | References |
|---|---|---|---|---|---|---|
| Pembrolizumab | PD-1 | 23 May 2017 | Patients with dMMR or MSI-H tumors | Pooled analysis on 149 patients enrolled across five single-arm studies. ORR: 39.6% (31.7–47.9%; 95% CI); DOR ≥ 6 months in 78% of patients. | pain in muscles, rash, diarrhea, fever, cough, decreased appetite, itching, shortness of breath, constipation, bones or joints and abdominal pain, nausea, and hypothyroidism | [38] |
| Larotrectinib | NTRK | 26 November 2018 | Patients with NTRK fusion-positive tumors | Pooled analysis on 55 patients enrolled across three single-arm studies. ORR: 75% (61–85%; 95% CI); 1-y PFS: 55%. | fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea | [63] |
| Entrectinib | NTRK | 15 August 2019 | Patients with NTRK fusion-positive tumors | Pooled analysis on 54 patients enrolled across three single-arm studies. ORR: 57% (43.2–70.8%; 95% CI); mDOR: 10 months (7.1-not estimable; 95% CI). | fatigue, constipation, dysgeusia, edema, dizziness, diarrhea, nausea, dysesthesia, dyspnea, myalgia, cognitive impairment, increased weight, cough, vomiting, pyrexia, arthralgia, and vision disorders | [66] |
| Pembrolizumab | PD-1 | 16 June 2020 | Patients with TMB-H tumors | Subgroup prespecified analysis from a multi-cohort single-arm phase II study. ORR: 29% (21–39%; 95% CI); mDOR: not reached (range 22–34.8 months). | pain in muscles, rash, diarrhea, fever, cough, decreased appetite, itching, shortness of breath, constipation, bones or joints and stomach-area (abdominal) pain, nausea, and low levels of thyroid hormone | [60] |
| Dostarlimab | PD-1 | 17 August 2021 | Patients with dMMR or MSI-H tumors | Prespecified interim analysis from a single-arm multi-cohort phase I study. ORR: 41.6% (34.9–48.6%; 95% CI); mDOR: 34.7 months (range 2.6–35.8). | fatigue/asthenia, anemia, rash, nausea, diarrhea, constipation, and vomiting | [46] |
| Dabrafenib + Trametinib | BRAF + MEK | 22 June 2022 | Patients with BRAFV600E mutated tumors | Pooled analysis on 167 patients (131 adults, 36 children) enrolled across three single-arm studies. ORR adults: 41% (33–50%; 95% CI); ORR children: 25% (12–42%; 95% CI). | pyrexia, fatigue, chills, peripheral edema, nausea, constipation, vomiting, diarrhea, rash, headache, hemorrhage, cough, myalgia, and arthralgia | [71] |
| Selpercatinib | RET | 21 September 2022 | Patients with RET fusion-positive tumors | Prespecified interim analysis from a multi-cohort single-arm phase I/II study. ORR: 43.9% (28.5–60.3%; 95% CI); mDOR: 24.5 months (9.2-not evaluable; 95% CI). | hypertension, prolonged QT interval, diarrhea, dyspnea, fatigue, abdominal pain, hemorrhage, headache, rash, constipation, nausea, vomiting, and edema | [82] |
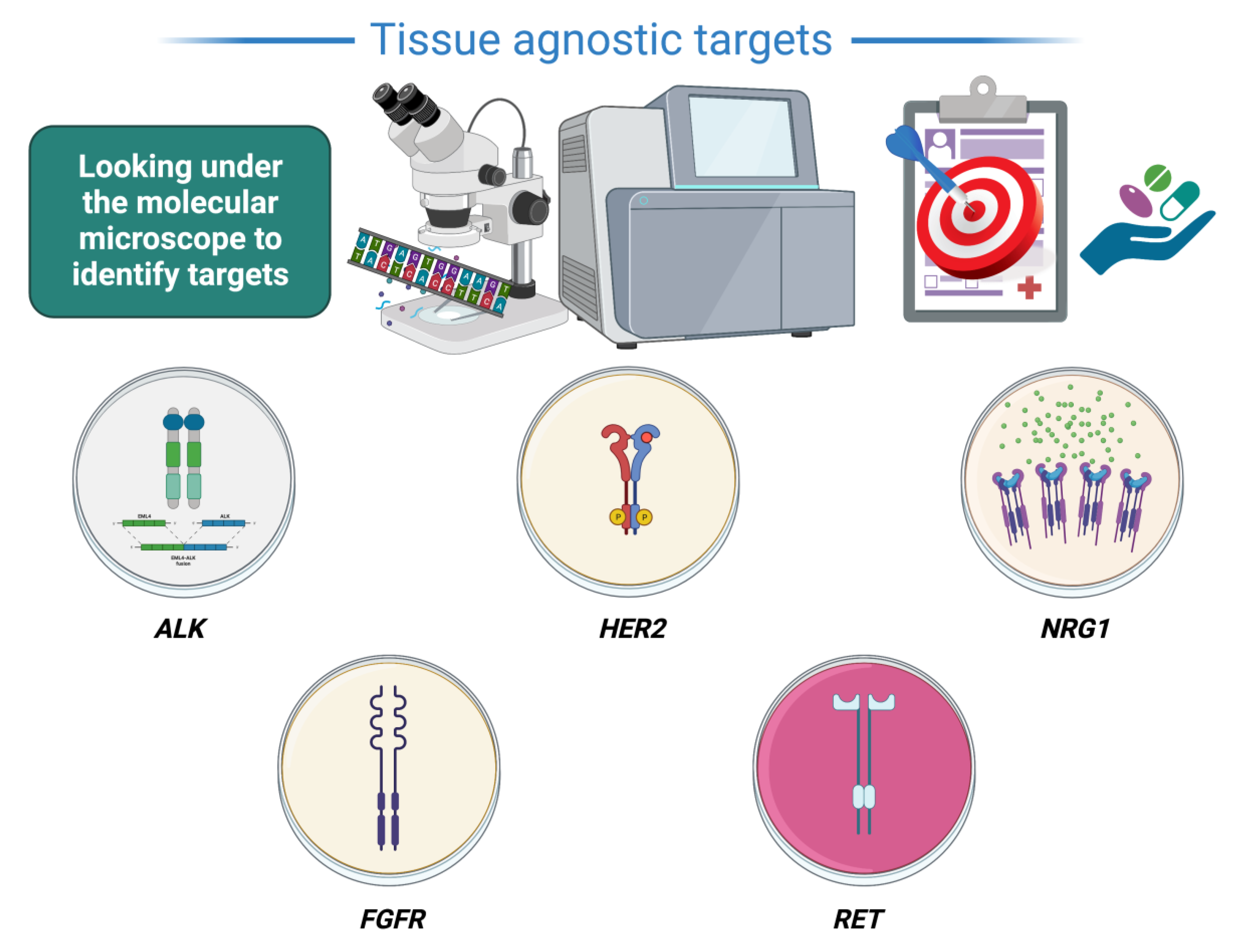
3. Agnostic Treatments Currently under Evaluation (Figure 1)
3.1. ALK

3.2. HER2
3.3. NRG1
3.4. FGFR
3.5. RET
3.6. Combinations N-of-1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hajdu, S.I. A Note from History: Landmarks in History of Cancer, Part 3. Cancer 2012, 118, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, S.I.; Vadmal, M. A Note from History: Landmarks in History of Cancer, Part 6. Cancer 2013, 119, 4058–4082. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, S.I.; Vadmal, M.; Tang, P. A Note from History: Landmarks in History of Cancer, Part 7. Cancer 2015, 121, 2480–2513. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Doroshow, J.H. Genomics and the History of Precision Oncology. Surg. Oncol. Clin. N. Am. 2020, 29, 35–49. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021, 7, 15–28. [Google Scholar] [CrossRef]
- Redig, A.J.; Jänne, P.A. Basket Trials and the Evolution of Clinical Trial Design in an Era of Genomic Medicine. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 975–977. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.J.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Sikalidis, A.K. Amino Acids and Immune Response: A Role for Cysteine, Glutamine, Phenylalanine, Tryptophan and Arginine in T-Cell Function and Cancer? Pathol. Oncol. Res. 2015, 21, 9–17. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, Leucovorin, and Oxaliplatin with and without Cetuximab in the First-Line Treatment of Metastatic Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized Medicine in a Phase I Clinical Trials Program: The MD Anderson Cancer Center Initiative. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Hong, D.S.; Ye, Y.; Cartwright, C.; Wheler, J.J.; Falchook, G.S.; Naing, A.; Fu, S.; Piha-Paul, S.; Janku, F.; et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. 2017, 1, 1–18. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular Profiling of Cancer Patients Enables Personalized Combination Therapy: The I-PREDICT Study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Patel, H.; Nikanjam, M.; Fanta, P.T.; Hahn, M.E.; De, P.; Williams, C.; Guido, J.; et al. Molecular Profiling of Advanced Malignancies Guides First-Line N-of-1 Treatments in the I-PREDICT Treatment-Naïve Study. Genome Med. 2021, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and Transcriptomic Profiling Expands Precision Cancer Medicine: The WINTHER Trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Tabernero, J. Optimal Design of Trials to Demonstrate the Utility of Genomically-Guided Therapy: Putting Precision Cancer Medicine to the Test. Mol. Oncol. 2015, 9, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Bogin, V. Master Protocols: New Directions in Drug Discovery. Contemp. Clin. trials Commun. 2020, 18, 100568. [Google Scholar] [CrossRef]
- FDA Modernizes Clinical Trials with Master Protocols. Available online: https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/fda-modernizes-clinical-trials-master-protocols (accessed on 4 December 2022).
- FDA Takes Steps to Provide Clarity on Developing New Drug Products in the Age of Individualized Medicine. Available online: https://www.fda.gov/news-events/press-announcements/fda-takes-steps-provide-clarity-developing-new-drug-products-age-individualized-medicine (accessed on 4 December 2022).
- Fountzilas, E.; Tsimberidou, A.M.; Vo, H.H.; Kurzrock, R. Clinical Trial Design in the Era of Precision Medicine. Genome Med. 2022, 14, 101. [Google Scholar] [CrossRef]
- Shaya, J.; Kato, S.; Adashek, J.J.; Patel, H.; Fanta, P.T.; Botta, G.P.; Sicklick, J.K.; Kurzrock, R. Personalized Matched Targeted Therapy in Advanced Pancreatic Cancer: A Pilot Cohort Analysis. NPJ Genom. Med. 2023, 8, 1. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncol. Annu. Meet. 2018, 38, 184–187. [Google Scholar] [CrossRef]
- Simon, R.; Roychowdhury, S. Implementing Personalized Cancer Genomics in Clinical Trials. Nat. Rev. Drug Discov. 2013, 12, 358–369. [Google Scholar] [CrossRef]
- Stewart, D.J.; Kurzrock, R. Fool’s Gold, Lost Treasures, and the Randomized Clinical Trial. BMC Cancer 2013, 13, 193. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Olave, M.C.; Graham, R.P. Mismatch Repair Deficiency: The What, How and Why It Is Important. Genes Chromosomes Cancer 2022, 61, 314–321. [Google Scholar] [CrossRef]
- Poole, R.M. Pembrolizumab: First Global Approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 19 November 2022).
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and Implications of FDA Approval of Pembrolizumab for Biomarker-Defined Disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef]
- Merck & Co. KEYTRUDA (Pembrolizumab) Prescribing Information. Available online: http://www.merck.com/product/usa/pi_circulars/k/%0Akeytruda/keytruda_pi.pdf (accessed on 19 November 2022).
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; De Jesus Acosta, A.; Doi, T.; Longo, F.; et al. Pembrolizumab in Microsatellite Instability High or Mismatch Repair Deficient Cancers: Updated Analysis from the Phase II KEYNOTE-158 Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H.J.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability-High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ghosh, S.; Sharma, G.; Wang, Z.; Kehry, M.R.; Marino, M.H.; Neben, T.Y.; Lu, S.; Luo, S.; Roberts, S.; et al. Preclinical Characterization of Dostarlimab, a Therapeutic Anti-PD-1 Antibody with Potent Activity to Enhance Immune Function in in Vitro Cellular Assays and in Vivo Animal Models. Mabs 2021, 13, 1954136. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Dostarlimab-Gxly for dMMR Advanced Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors (accessed on 19 November 2022).
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.-P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef]
- Patnaik, A.; Weiss, G.J.; Rasco, D.W.; Blaydorn, L.; Mirabella, A.; Beeram, M.; Guo, W.; Lu, S.; Danaee, H.; McEachern, K.; et al. Safety, Antitumor Activity, and Pharmacokinetics of Dostarlimab, an Anti-PD-1, in Patients with Advanced Solid Tumors: A Dose-Escalation Phase 1 Trial. Cancer Chemother. Pharmacol. 2022, 89, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Roda, D.; Pikiel, J.; Trigo, J.; Bosch-Barrera, J.; Drew, Y.; Kristeleit, R.; Hiret, S.; Bajor, D.L.; Cruz, P.; et al. Safety and Efficacy of Dostarlimab in Patients With Recurrent/Advanced Non-Small Cell Lung Cancer: Results from Cohort E of the Phase I GARNET Trial. Clin. Lung Cancer 2022, 23, e415–e427. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’Malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and Antitumor Activity of Dostarlimab in Patients with Advanced or Recurrent DNA Mismatch Repair Deficient/Microsatellite Instability-High (DMMR/MSI-H) or Proficient/Stable (MMRp/MSS) Endometrial Cancer: Interim Results from GARNET-a Phase I, Sing. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef]
- Oaknin, A.; Bosse, T.J.; Creutzberg, C.L.; Giornelli, G.; Harter, P.; Joly, F.; Lorusso, D.; Marth, C.; Makker, V.; Mirza, M.R.; et al. ESMO Guidelines Committee. Ann. Oncol. 2022, 33 (Suppl. S7), S235–S282. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Ellard, S.; Trigo Pérez, J.M.; Arkenau, H.-T.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and Efficacy of Anti–PD-1 Antibody Dostarlimab in Patients (Pts) with Mismatch Repair-Deficient (DMMR) Solid Cancers: Results from GARNET Study. J. Clin. Oncol. 2021, 39 (Suppl. S3), 9. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Jimenez-Rodriguez, B.; Ellard, S.; Gravina, A.; Miller, R.; Tinker, A.; Jewell, A.; Pikiel, J.; et al. Efficacy and Safety of Dostarlimab in Patients (Pts) with Mismatch Repair Deficient (DMMR) Solid Tumors: Analysis of 2 Cohorts in the GARNET Study. J. Clin. Oncol. 2022, 40 (Suppl. S16), 2587. [Google Scholar] [CrossRef]
- Berton, D.; Banerjee, S.N.; Curigliano, G.; Cresta, S.; Arkenau, H.-T.; Abdeddaim, C.; Kristeleit, R.S.; Redondo, A.; Leath, C.A.; Antón Torres, A.; et al. Antitumor Activity of Dostarlimab in Patients with Mismatch Repair-Deficient/Microsatellite Instability–High Tumors: A Combined Analysis of Two Cohorts in the GARNET Study. J. Clin. Oncol. 2021, 39 (Suppl. S15), 2564. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 19 November 2022).
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.J.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet. Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK Family Proteins in Cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef]
- FDA Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions (accessed on 20 November 2022).
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 21 November 2022).
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet. Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Demetri, G.D.; De Braud, F.; Drilon, A.; Siena, S.; Patel, M.R.; Cho, B.C.; Liu, S.V.; Ahn, M.-J.; Chiu, C.-H.; Lin, J.J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Patients with NTRK Fusion-Positive Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1302–1312. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Adashek, J.J.; Menta, A.K.; Reddy, N.K.; Desai, A.P.; Roszik, J.; Subbiah, V. Tissue-Agnostic Activity of BRAF plus MEK Inhibitor in BRAF V600-Mutant Tumors. Mol. Cancer Ther. 2022, 21, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Dabrafenib in Combination with Trametinib for Unresectable or Metastatic Solid Tumors with BRAF V600E Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid (accessed on 22 November 2022).
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.J.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus Trametinib in Patients with Previously Untreated BRAF(V600E)-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.-I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Stein, A.; van den Bent, M.; De Greve, J.; Wick, A.; de Vos, F.Y.F.L.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; et al. Dabrafenib plus Trametinib in Patients with BRAF(V600E)-Mutant Low-Grade and High-Grade Glioma (ROAR): A Multicentre, Open-Label, Single-Arm, Phase 2, Basket Trial. Lancet Oncol. 2022, 23, 53–64. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF(V600E)-Mutated Biliary Tract Cancer (ROAR): A Phase 2, Open-Label, Single-Arm, Multicentre Basket Trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF V600E-Mutant Anaplastic Thyroid Cancer: Updated Analysis from the Phase II ROAR Basket Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET Fusions in Solid Tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Kurzrock, R. Selpercatinib Aimed at RET-Altered Cancers. N. Engl. J. Med. 2020, 383, 868–869. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Selpercatinib for Locally Advanced or Metastatic RET Fusion-Positive Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-solid-tumors (accessed on 26 November 2022).
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-Agnostic Efficacy and Safety of Selpercatinib in Patients with RET Fusion-Positive Solid Tumours Other than Lung or Thyroid Tumours (LIBRETTO-001): A Phase 1/2, Open-Label, Basket Trial. Lancet. Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Dvorak, C.; Henkle, C.; Ellingham, T.; Perlman, E.J. Recurrent Involvement of 2p23 in Inflammatory Myofibroblastic Tumors. Cancer Res. 1999, 59, 2776–2780. [Google Scholar] [PubMed]
- Lawrence, B.; Perez-Atayde, A.; Hibbard, M.K.; Rubin, B.P.; Dal Cin, P.; Pinkus, J.L.; Pinkus, G.S.; Xiao, S.; Yi, E.S.; Fletcher, C.D.; et al. TPM3-ALK and TPM4-ALK Oncogenes in Inflammatory Myofibroblastic Tumors. Am. J. Pathol. 2000, 157, 377–384. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Pillay, K.; Govender, D.; Chetty, R. ALK Protein Expression in Rhabdomyosarcomas. Histopathology 2002, 41, 461–467. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of Neuroblastoma Identifies Chromothripsis and Defects in Neuritogenesis Genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. Mechanistic Insight into ALK Receptor Tyrosine Kinase in Human Cancer Biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The Role of the ALK Receptor in Cancer Biology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 (Suppl. S3), iii4–iii15. [Google Scholar] [CrossRef]
- Ross, J.S.; Ali, S.M.; Fasan, O.; Block, J.; Pal, S.; Elvin, J.A.; Schrock, A.B.; Suh, J.; Nozad, S.; Kim, S.; et al. ALK Fusions in a Wide Variety of Tumor Types Respond to Anti-ALK Targeted Therapy. Oncologist 2017, 22, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Ali, S.M.; Lacy, J.; Hendifar, A.; Nguyen, K.; Koo, J.; Chung, J.H.; Greenbowe, J.; Ross, J.S.; Nikiforova, M.N.; et al. Identification of Targetable ALK Rearrangements in Pancreatic Ductal Adenocarcinoma. J. Natl. Compr. Canc. Netw. 2017, 15, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Salgia, S.K.; Govindarajan, A.; Salgia, R.; Pal, S.K. ALK-Directed Therapy in Non-NSCLC Malignancies: Are We Ready? JCO Precis. Oncol. 2021, 5, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Li, Y.Y.; Spurr, L.F.; Shinagare, A.B.; Abhyankar, R.; Reilly, E.; Brais, L.K.; Nag, A.; Ducar, M.D.; Thorner, A.R.; et al. Molecular Characterization and Therapeutic Targeting of Colorectal Cancers Harboring Receptor Tyrosine Kinase Fusions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.J.; Kim, H.R.; Shin, E.; Kim, C.; Heo, S.J.; Lee, C.-K.; Park, J.K.; Noh, S.H.; Chung, H.C.; Rha, S.Y. The Clinicopathologic Features and Prognostic Impact of ALK Positivity in Patients with Resected Gastric Cancer. Ann. Surg. Oncol. 2015, 22, 3938–3945. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. ALKoma: A Cancer Subtype with a Shared Target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.-S.; Beck, J.T.; Edenfield, W.J.; et al. Long-Term Effects of Crizotinib in ALK-Positive Tumors (Excluding NSCLC): A Phase 1b Open-Label Study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.-C.; Gadgeel, S.M.; et al. Lorlatinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer: Results from a Global Phase 2 Study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Schöffski, P.; Sufliarsky, J.; Gelderblom, H.; Blay, J.-Y.; Strauss, S.J.; Stacchiotti, S.; Rutkowski, P.; Lindner, L.H.; Leahy, M.G.; Italiano, A.; et al. Crizotinib in Patients with Advanced, Inoperable Inflammatory Myofibroblastic Tumours with and without Anaplastic Lymphoma Kinase Gene Alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): A Multicentre, Single-Drug, Pros. Lancet Respir. Med. 2018, 6, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Ohyanagi, F.; Ariyasu, R.; Koyama, J.; Sonoda, T.; Nishikawa, S.; Kitazono, S.; Yanagitani, N.; Horiike, A.; Ninomiya, H.; et al. Dramatic Response to Alectinib in Inflammatory Myofibroblastic Tumor with Anaplastic Lymphoma Kinase Fusion Gene. Jpn. J. Clin. Oncol. 2017, 47, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Bergerot, P.; Dizman, N.; Bergerot, C.; Adashek, J.; Madison, R.; Chung, J.H.; Ali, S.M.; Jones, J.O.; Salgia, R. Responses to Alectinib in ALK-Rearranged Papillary Renal Cell Carcinoma. Eur. Urol. 2018, 74, 124–128. [Google Scholar] [CrossRef]
- Rao, N.; Iwenofu, H.; Tang, B.; Woyach, J.; Liebner, D.A. Inflammatory Myofibroblastic Tumor Driven by Novel NUMA1-ALK Fusion Responds to ALK Inhibition. J. Natl. Compr. Canc. Netw. 2018, 16, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Kadowaki, S.; Kato, K.; Hanai, N.; Hasegawa, Y.; Yatabe, Y.; Muro, K. Durable Response to the ALK Inhibitor Alectinib in Inflammatory Myofibroblastic Tumor of the Head and Neck with a Novel SQSTM1-ALK Fusion: A Case Report. Investig. New Drugs 2019, 37, 791–795. [Google Scholar] [CrossRef]
- Takeyasu, Y.; Okuma, H.S.; Kojima, Y.; Nishikawa, T.; Tanioka, M.; Sudo, K.; Shimoi, T.; Noguchi, E.; Arakawa, A.; Mori, T.; et al. Impact of ALK Inhibitors in Patients With ALK-Rearranged Nonlung Solid Tumors. JCO Precis. Oncol. 2021, 5, 756–766. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic Non-Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29 (Suppl. S4), iv192–iv237. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 2.2021. J. Natl. Compr. Canc. Netw. 2021, 19, 254–266. [Google Scholar] [CrossRef]
- Ambrosini, M.; Del Re, M.; Manca, P.; Hendifar, A.; Drilon, A.; Harada, G.; Ree, A.H.; Klempner, S.; Mælandsmo, G.M.; Flatmark, K.; et al. ALK Inhibitors in Patients with ALK Fusion-Positive GI Cancers: An International Data Set and a Molecular Case Series. JCO Precis. Oncol. 2022, 6, e2200015. [Google Scholar] [CrossRef]
- Adashek, J.J.; Sapkota, S.; de Castro Luna, R.; Seiwert, T.Y. Complete Response to Alectinib in ALK-Fusion Metastatic Salivary Ductal Carcinoma. NPJ Precis. Oncol. 2023, 7, 36. [Google Scholar] [CrossRef]
- Corti, C.; Giugliano, F.; Nicolò, E.; Ascione, L.; Curigliano, G. Antibody-Drug Conjugates for the Treatment of Breast Cancer. Cancers 2021, 13, 2898. [Google Scholar] [CrossRef] [PubMed]
- Gennari, A.; André, F.; Barrios, C.H.; Cortés, J.; de Azambuja, E.; DeMichele, A.; Dent, R.; Fenlon, D.; Gligorov, J.; Hurvitz, S.A.; et al. ESMO Clinical Practice Guideline for the Diagnosis, Staging and Treatment of Patients with Metastatic Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef] [PubMed]
- Corti, C.; Antonarelli, G.; Valenza, C.; Nicolò, E.; Rugo, H.; Cortés, J.; Harbeck, N.; Carey, L.A.; Criscitiello, C.; Curigliano, G. Histology-Agnostic Approvals for Antibody-Drug Conjugates in Solid Tumours: Is the Time Ripe? Eur. J. Cancer 2022, 171, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Summaries of Safety Labeling Changes Approved by FDA-Boxed Warnings Highlights, July-September 2022. Am. J. Heal. Pharm. AJHP Off. J. Am. Soc. Health Pharm. 2022, 79, e157–e160. [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in Combination with Chemotherapy versus Chemotherapy Alone for Treatment of HER2-Positive Advanced Gastric or Gastro-Oesophageal Junction Cancer (ToGA): A Phase 3, Open-Label, Randomised Controlled Trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Tabernero, J.; Hoff, P.M.; Shen, L.; Ohtsu, A.; Shah, M.A.; Cheng, K.; Song, C.; Wu, H.; Eng-Wong, J.; Kim, K.; et al. Pertuzumab plus Trastuzumab and Chemotherapy for HER2-Positive Metastatic Gastric or Gastro-Oesophageal Junction Cancer (JACOB): Final Analysis of a Double-Blind, Randomised, Placebo-Controlled Phase 3 Study. Lancet Oncol. 2018, 19, 1372–1384. [Google Scholar] [CrossRef]
- Thuss-Patience, P.C.; Shah, M.A.; Ohtsu, A.; Van Cutsem, E.; Ajani, J.A.; Castro, H.; Mansoor, W.; Chung, H.C.; Bodoky, G.; Shitara, K.; et al. Trastuzumab Emtansine versus Taxane Use for Previously Treated HER2-Positive Locally Advanced or Metastatic Gastric or Gastro-Oesophageal Junction Adenocarcinoma (GATSBY): An International Randomised, Open-Label, Adaptive, Phase 2/3 Study. Lancet Oncol. 2017, 18, 640–653. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-Targeted Therapy with Trastuzumab and Lapatinib in Treatment-Refractory, KRAS Codon 12/13 Wild-Type, HER2-Positive Metastatic Colorectal Cancer (HERACLES): A Proof-of-Concept, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Lonardi, S.; Martino, C.; Fenocchio, E.; Tosi, F.; Ghezzi, S.; Leone, F.; Bergamo, F.; Zagonel, V.; Ciardiello, F.; et al. Pertuzumab and Trastuzumab Emtansine in Patients with HER2-Amplified Metastatic Colorectal Cancer: The Phase II HERACLES-B Trial. ESMO Open 2020, 5, e000911. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and Trastuzumab for HER2-Positive, Metastatic Biliary Tract Cancer (MyPathway): A Multicentre, Open-Label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Mazières, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse, B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung Cancer That Harbors an HER2 Mutation: Epidemiologic Characteristics and Therapeutic Perspectives. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Peters, S. Targeting HER2-Mutant NSCLC—The Light Is On. N. Engl. J. Med. 2022, 386, 286–289. [Google Scholar] [CrossRef]
- Rosas, D.; Raez, L.E.; Russo, A.; Rolfo, C. Neuregulin 1 Gene (NRG1). A Potentially New Targetable Alteration for the Treatment of Lung Cancer. Cancers 2021, 13, 5038. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Rossi, A. NRG1: A Cinderella Fusion in Lung Cancer? Lung Cancer Manag. 2017, 6, 121–123. [Google Scholar] [CrossRef]
- Trombetta, D.; Rossi, A.; Fabrizio, F.P.; Sparaneo, A.; Graziano, P.; Fazio, V.M.; Muscarella, L.A. NRG1-ErbB Lost in Translation: A New Paradigm for Lung Cancer? Curr. Med. Chem. 2017, 24, 4213–4228. [Google Scholar] [CrossRef]
- Schaefer, G.; Fitzpatrick, V.D.; Sliwkowski, M.X. Gamma-Heregulin: A Novel Heregulin Isoform That Is an Autocrine Growth Factor for the Human Breast Cancer Cell Line, MDA-MB-175. Oncogene 1997, 15, 1385–1394. [Google Scholar] [CrossRef]
- Jonna, S.; Feldman, R.A.; Swensen, J.; Gatalica, Z.; Korn, W.M.; Borghaei, H.; Ma, P.C.; Nieva, J.J.; Spira, A.I.; Vanderwalde, A.M.; et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4966–4972. [Google Scholar] [CrossRef]
- Nagasaka, M.; Ou, S.-H.I. NRG1 and NRG2 Fusion Positive Solid Tumor Malignancies: A Paradigm of Ligand-Fusion Oncogenesis. Trends Cancer 2022, 8, 242–258. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Xiu, J.; Nagasaka, M.; Xia, B.; Zhang, S.S.; Zhang, Q.; Swensen, J.J.; Spetzler, D.; Korn, W.M.; Zhu, V.W.; et al. Identification of Novel CDH1-NRG2α and F11R-NRG2α Fusions in NSCLC Plus Additional Novel NRG2α Fusions in Other Solid Tumors by Whole Transcriptome Sequencing. JTO Clin. Res. Rep. 2021, 2, 100132. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Goto, K.; Kim, D.-W.; Martin-Romano, P.; Ou, S.-H.I.; O’Kane, G.M.; O’Reilly, E.M.; Umemoto, K.; Duruisseaux, M.; Neuzillet, C.; et al. Efficacy and Safety of Zenocutuzumab, a HER2 x HER3 Bispecific Antibody, across Advanced NRG1 Fusion (NRG1+) Cancers. J. Clin. Oncol. 2022, 40 (Suppl. S16), 105. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Li, I.W.; Krishnamurthy, N.; Wei, G.; Li, G. Opportunities and Challenges in Developing Tissue-Agnostic Anti-Cancer Drugs. J. Cancer Metastasis Treat. 2020, 6, 14. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast Growth Factor Receptor 2 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive Parallel Sequencing Uncovers Actionable FGFR2-PPHLN1 Fusion and ARAF Mutations in Intrahepatic Cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR Family Gene Fusions as Therapeutic Targets in a Wide Spectrum of Solid Tumours. J. Pathol. 2014, 232, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Hollebecque, A.; Borad, M.; Sahai, V.; Catenacci, D.V.T.; Murphy, A.; Vaccaro, G.; Paulson, A.; Oh, D.-Y.; Féliz, L.; Lihou, C.; et al. Interim Results of Fight-202, a Phase II, Open-Label, Multicenter Study of INCB054828 in Patients (Pts) with Previously Treated Advanced/Metastatic or Surgically Unresectable Cholangiocarcinoma (CCA) with/without Fibroblast Growth Factor (FGF)/FGF Recepto. Ann. Oncol. 2018, 29, viii258. [Google Scholar] [CrossRef]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef]
- Park, J.O.; Feng, Y.-H.; Chen, Y.-Y.; Su, W.-C.; Oh, D.-Y.; Shen, L.; Kim, K.-P.; Liu, X.; Bai, Y.; Liao, H.; et al. Updated Results of a Phase IIa Study to Evaluate the Clinical Efficacy and Safety of Erdafitinib in Asian Advanced Cholangiocarcinoma (CCA) Patients with FGFR Alterations. J. Clin. Oncol. 2019, 37 (Suppl. S15), 4117. [Google Scholar] [CrossRef]
- Massard, C.; Pant, S.; Iyer, G.; Schuler, M.H.; Witt, O.; Qin, S.; Tabernero, J.; Doi, T.; Hargrave, D.R.; Hammond, C.; et al. Preliminary Results of Molecular Screening for FGFR Alterations (Alts) in the RAGNAR Histology-Agnostic Study with the FGFR-Inhibitor (FGFRi) Erdafitinib. J. Clin. Oncol. 2021, 39 (Suppl. S15), 4081. [Google Scholar] [CrossRef]
- Loriot, Y.; Schuler, M.H.; Iyer, G.; Witt, O.; Doi, T.; Qin, S.; Tabernero, J.; Reardon, D.A.; Massard, C.; Palmer, D.; et al. Tumor Agnostic Efficacy and Safety of Erdafitinib in Patients (Pts) with Advanced Solid Tumors with Prespecified Fibroblast Growth Factor Receptor Alterations (FGFRalt) in RAGNAR: Interim Analysis (IA) Results. J. Clin. Oncol. 2022, 40 (Suppl. S16), 3007. [Google Scholar] [CrossRef]
- Gainor, J.F.; Curigliano, G.; Kim, D.-W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET Fusion-Positive Non-Small-Cell Lung Cancer (ARROW): A Multi-Cohort, Open-Label, Phase 1/2 Study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for Patients with Advanced or Metastatic RET-Altered Thyroid Cancer (ARROW): A Multi-Cohort, Open-Label, Registrational, Phase 1/2 Study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef]
- FDA Approves Pralsetinib for RET-Altered Thyroid Cancers. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pralsetinib-ret-altered-thyroid-cancers (accessed on 29 November 2022).
- FDA Approves Pralsetinib for Lung Cancer with RET Gene Fusions. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pralsetinib-lung-cancer-ret-gene-fusions (accessed on 29 November 2022).
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-Cancer Efficacy of Pralsetinib in Patients with RET Fusion-Positive Solid Tumors from the Phase 1/2 ARROW Trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef]
- Okamura, R.; Boichard, A.; Kato, S.; Sicklick, J.K.; Bazhenova, L.; Kurzrock, R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis. Oncol. 2018, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Westphalen, C.B.; Naing, A.; Kato, S.; Kurzrock, R. Cancer: Slaying the Nine-Headed Hydra. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 61–69. [Google Scholar] [CrossRef]
- Adashek, J.J.; Arroyo-Martinez, Y.; Menta, A.K.; Kurzrock, R.; Kato, S. Therapeutic Implications of Epidermal Growth Factor Receptor (EGFR) in the Treatment of Metastatic Gastric/GEJ Cancer. Front. Oncol. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S Phase Cell-Cycle Genomic Alterations and Accompanying Co-Alterations with Individualized CDK4/6 Inhibitor-Based Regimens. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Adashek, J.J.; Aguiar, P.N.; Castelo-Branco, P.; Lopes, G.L.J.; de Mello, R.A. PALOMA-3 Clinical Trial: Is There a Significant Benefit in Overall Survival for Breast Cancer? Is It Worth It? Future Oncol. 2019, 15, 1407–1410. [Google Scholar] [CrossRef]
- Adashek, J.J.; Goloubev, A.; Kato, S.; Kurzrock, R. Missing the Target in Cancer Therapy. Nat. Cancer 2021, 2, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Gumas, S.; Adashek, J.J.; Okamura, R.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Multi-Omic Analysis in Carcinoma of Unknown Primary (CUP): Therapeutic Impact of Knowing the Unknown. Mol. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-Analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tateo, V.; Marchese, P.V.; Mollica, V.; Massari, F.; Kurzrock, R.; Adashek, J.J. Agnostic Approvals in Oncology: Getting the Right Drug to the Right Patient with the Right Genomics. Pharmaceuticals 2023, 16, 614. https://doi.org/10.3390/ph16040614
Tateo V, Marchese PV, Mollica V, Massari F, Kurzrock R, Adashek JJ. Agnostic Approvals in Oncology: Getting the Right Drug to the Right Patient with the Right Genomics. Pharmaceuticals. 2023; 16(4):614. https://doi.org/10.3390/ph16040614
Chicago/Turabian StyleTateo, Valentina, Paola Valeria Marchese, Veronica Mollica, Francesco Massari, Razelle Kurzrock, and Jacob J. Adashek. 2023. "Agnostic Approvals in Oncology: Getting the Right Drug to the Right Patient with the Right Genomics" Pharmaceuticals 16, no. 4: 614. https://doi.org/10.3390/ph16040614