
Hidden Pool of Cardiac Adenine Nucleotides That Controls Adenosine Production
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
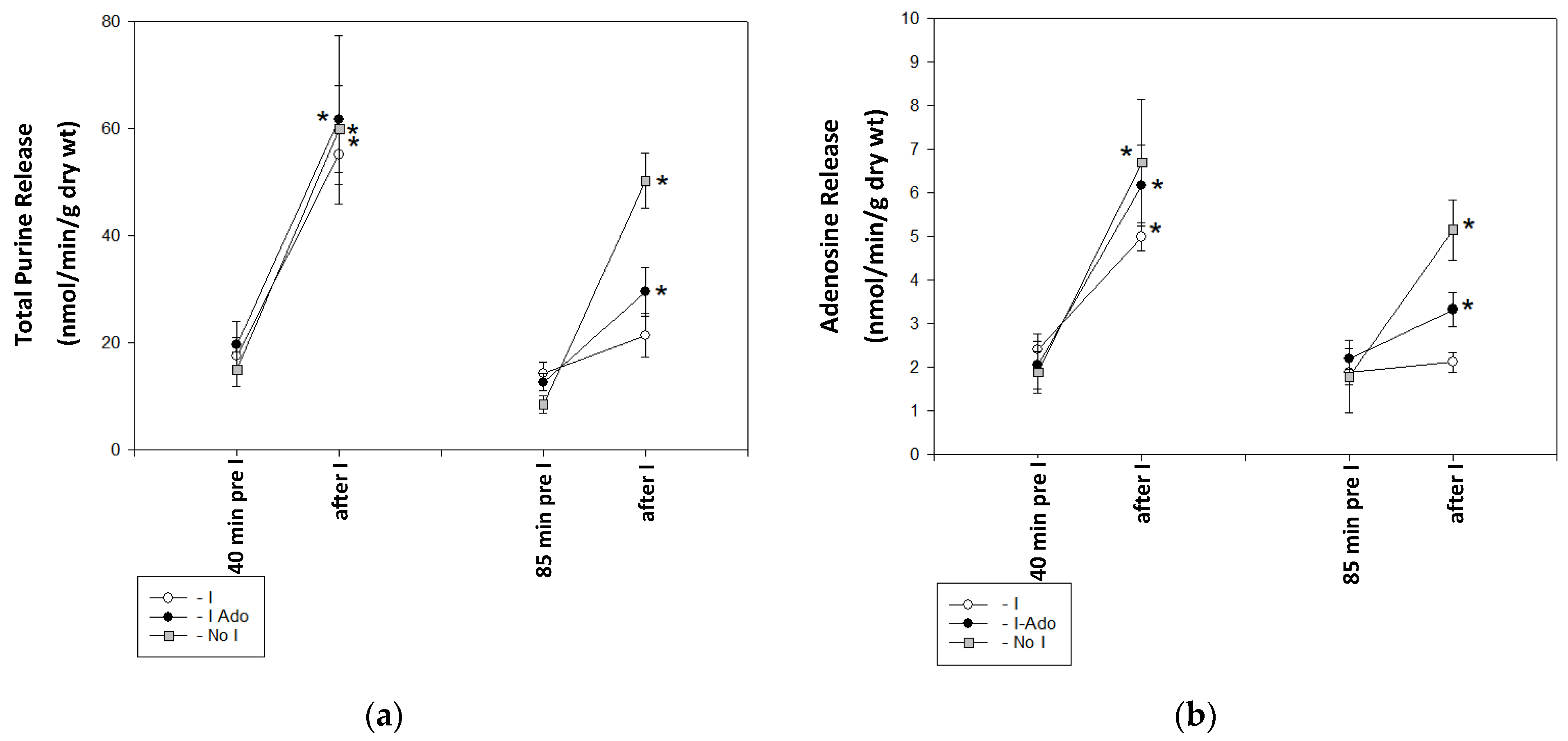
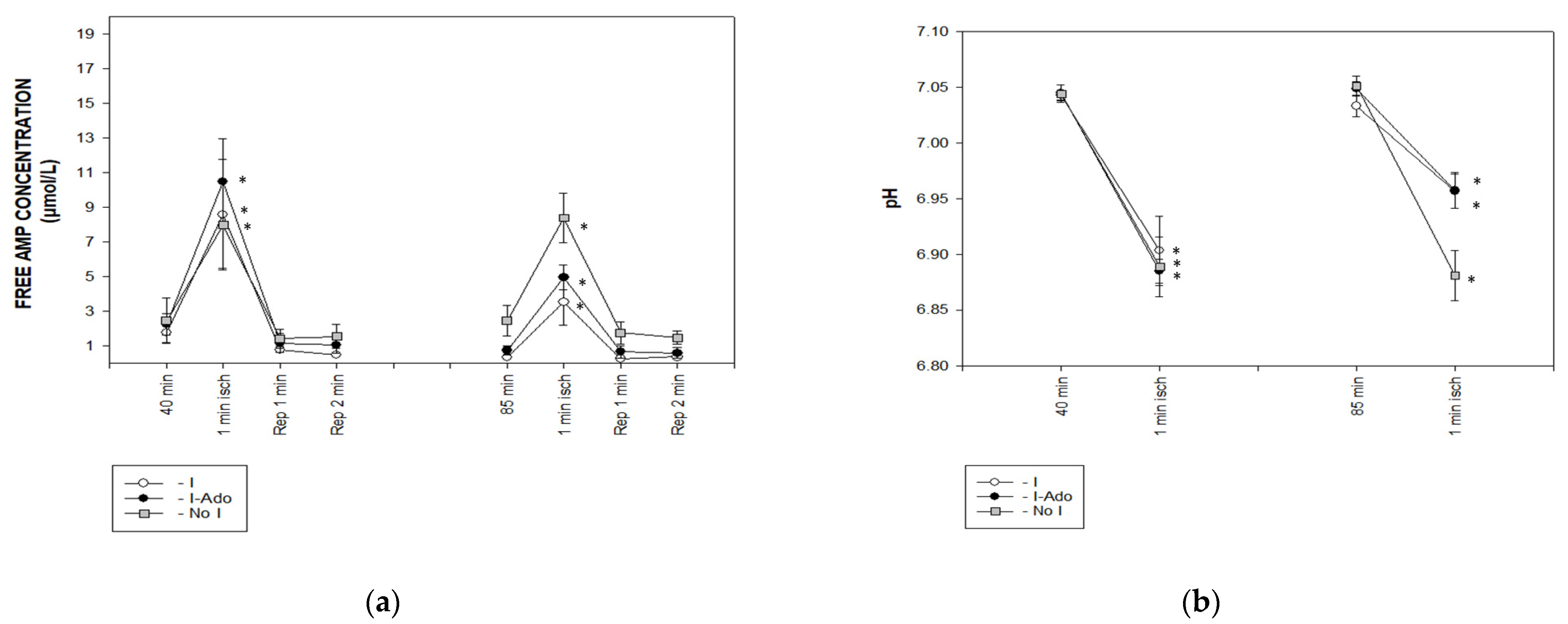
2.1. Release of Purine Catabolites and Adenosine in Coronary Effluent
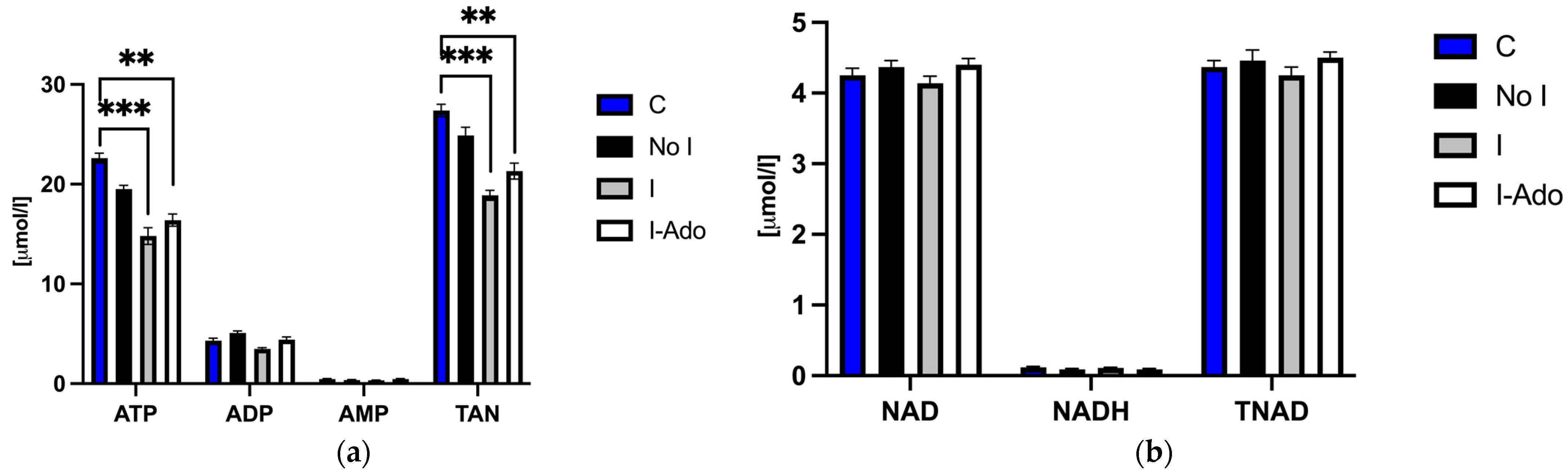
2.2. ATP (Adenosine Triphosphate), ADP (Adenosine Diphosphate), AMP (Adenosine Monophosphate), NAD (Nicotinamide Adenine Dinucleotide), Oxidized and Reduced Form of NAD (NADH), Total Adenine Nucleotide Pool (TAN), and Total NAD Pool (TNAD) in the Heart
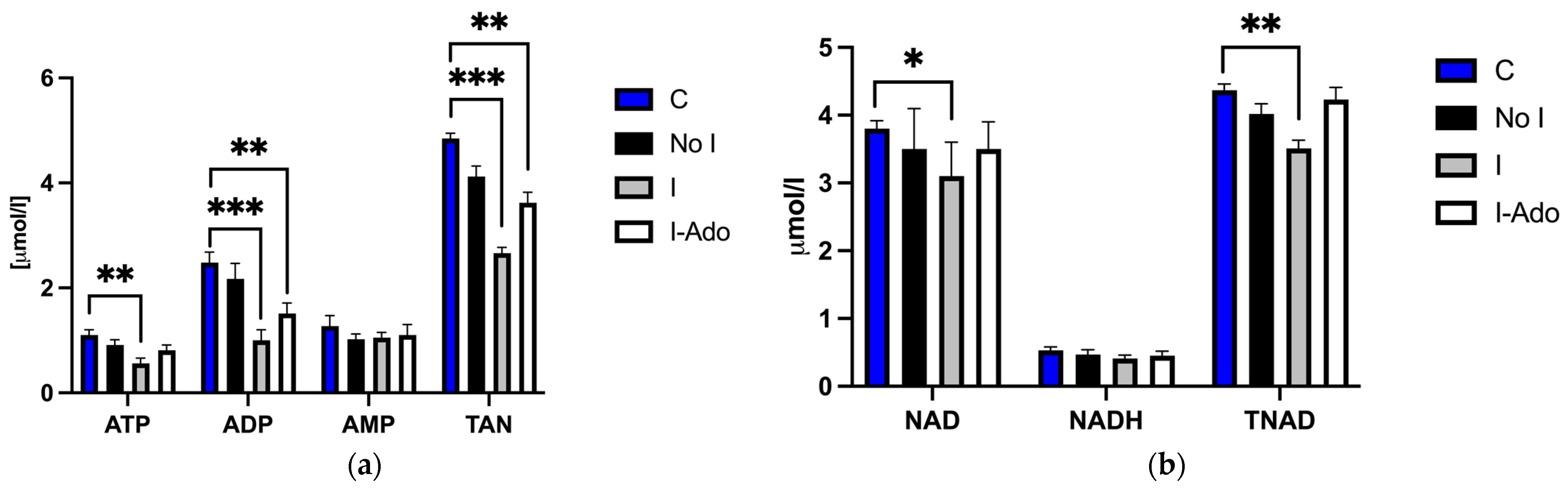
2.3. Mitochondrial ATP, ADP, AMP, NAD, NADH, TAN, and TNAD in the Heart
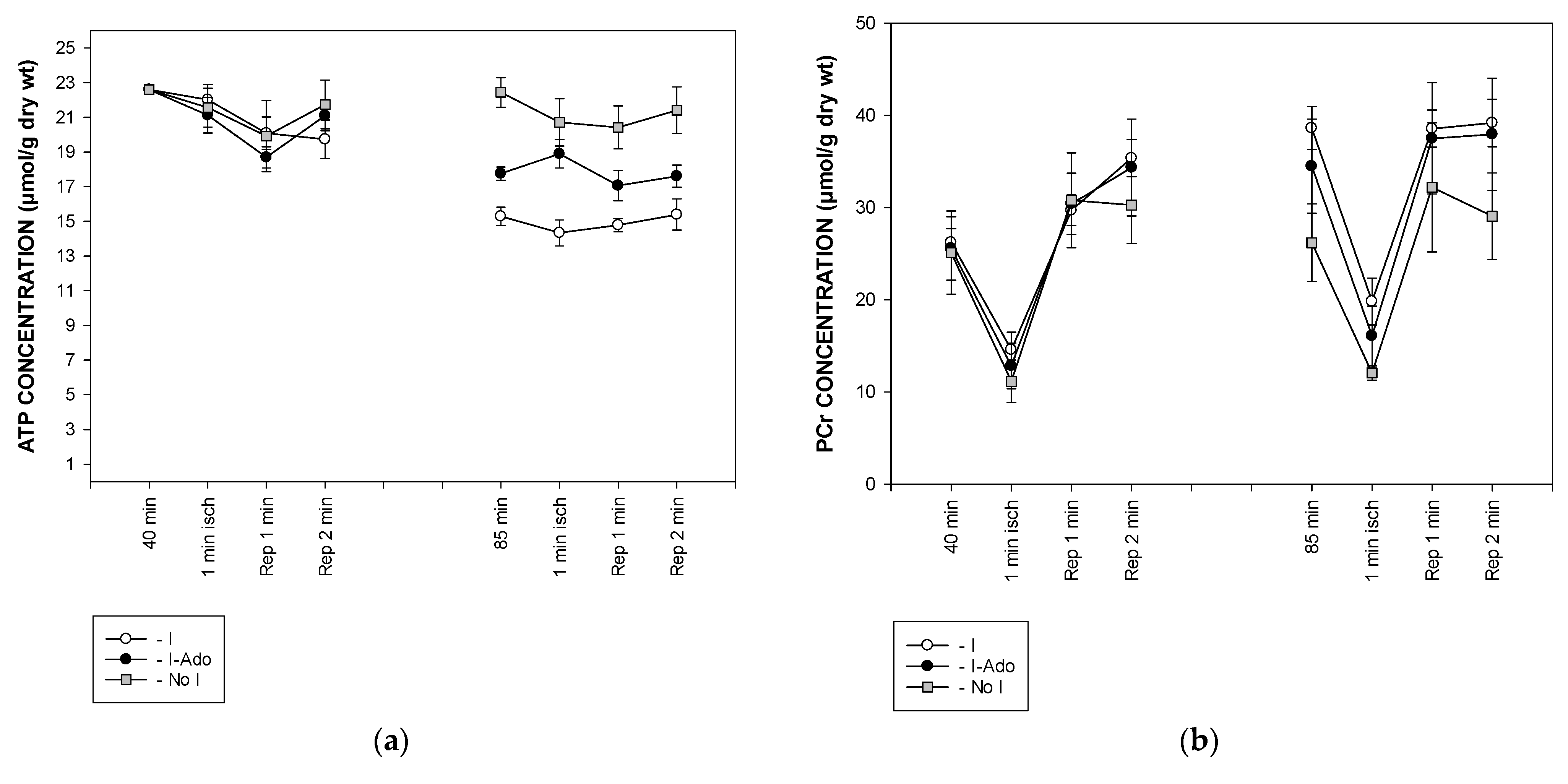
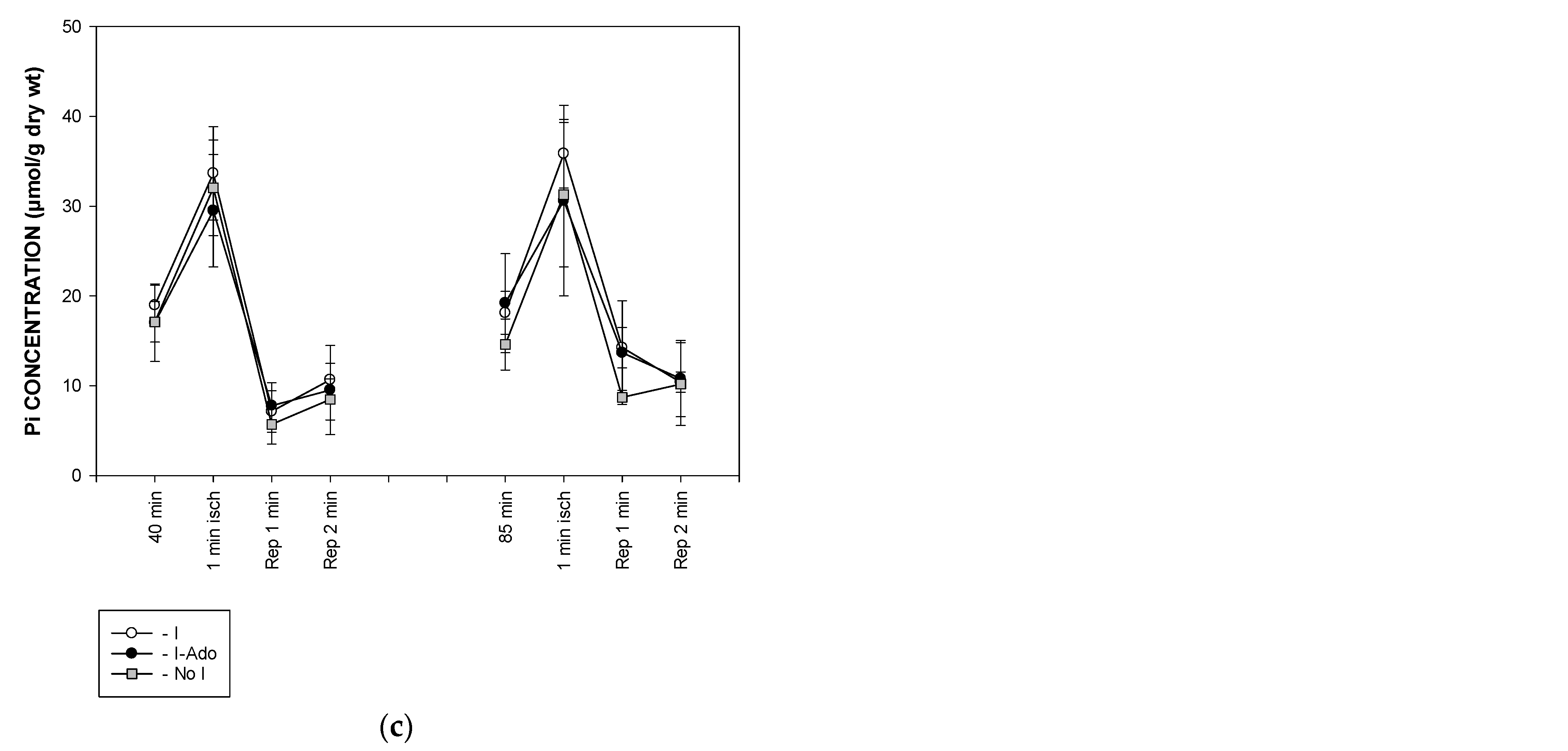
2.4. Concentration of Free AMP Calculated Based on 31P NMR (Nuclear Magnetic Resonance) Spectra
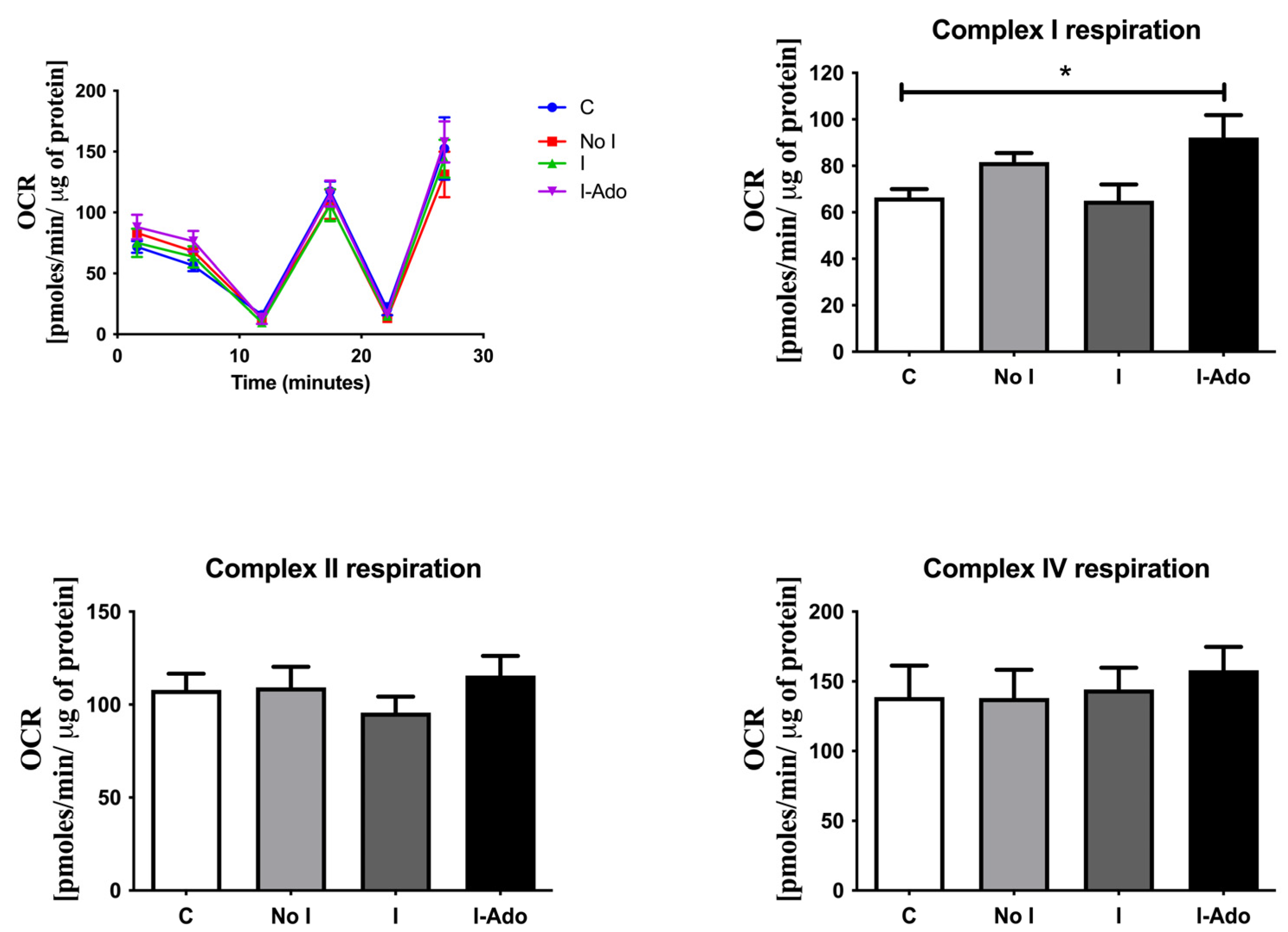
2.5. Oxygen Consumption in the Mitochondria of Rat Hearts
3. Discussion
4. Materials and Methods
4.1. Heart Perfusion
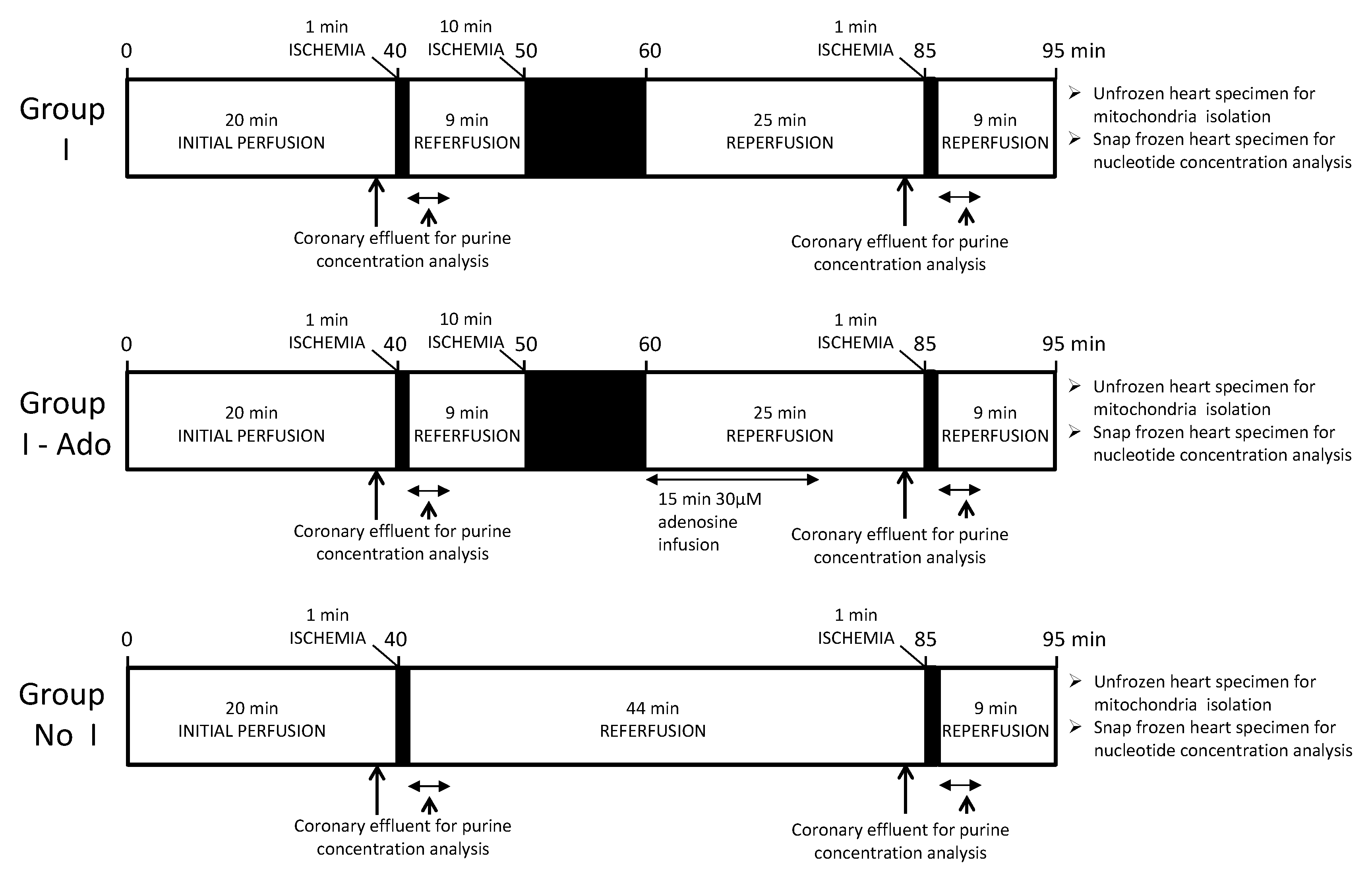
4.2. Experimental Protocol
4.3. Tissue Extraction for High-Performance Liquid Chromatography (HPLC) Analysis
4.4. 31P NMR Spectroscopy
4.5. Mitochondrial Respiration Analysis
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hori, M.; Kitakaze, M. Adenosine, the Heart, and Coronary Circulation. Hypertension 1991, 18, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, W.H.; Becker, B.F.; Heier, M.; Nees, S.; Gerlach, E. Endothelium-Mediated Coronary Dilatation by Adenosine Does Not Depend on Endothelial Adenylate Cyclase Activation: Studies in Isolated Guinea Pig Hearts. Pflug. Arch. 1988, 413, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nees, S.; Herzog, V.; Becker, B.F.; Bock, M.; Des Rosiers, C.; Gerlach, E. The Coronary Endothelium: A Highly Active Metabolic Barrier for Adenosine. Basic Res. Cardiol. 1985, 80, 515–529. [Google Scholar] [CrossRef]
- Mubagwa, K.; Mullane, K.; Flameng, W. Role of Adenosine in the Heart and Circulation. Cardiovasc. Res. 1996, 32, 797–813. [Google Scholar] [CrossRef] [Green Version]
- Hasko, G.; Cronstein, B.N. Adenosine: An Endogenous Regulator of Innate Immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, Inflammation and Cancer: A Leading Role for Adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Liem, D.A.; Verdouw, P.D.; Ploeg, H.; Kazim, S.; Duncker, D.J. Sites of Action of Adenosine in Interorgan Preconditioning of the Heart. Am. J. Physiol. Circ. Physiol. 2002, 283, H29–H37. [Google Scholar] [CrossRef] [Green Version]
- Ely, S.W.; Berne, R.M. Protective Effects of Adenosine in Myocardial Ischemia. Circulation 1992, 85, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Kroll, K.; Schrader, J.; Piper, H.M.; Henrich, M. Release of Adenosine and Cyclic AMP from Coronary Endothelium in Isolated Guinea Pig Hearts: Relation to Coronary Flow. Circ. Res. 1987, 60, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Mullane, K.; Bullough, D. Harnessing an Endogenous Cardioprotective Mechanism: Cellular Sources and Sites of Action of Adenosine. J. Mol. Cell. Cardiol. 1995, 27, 1041–1054. [Google Scholar] [CrossRef]
- Belardinelli, L.; Linden, J.; Berne, R.M. The Cardiac Effects of Adenosine. Prog. Cardiovasc. Dis. 1989, 32, 73–97. [Google Scholar] [CrossRef] [PubMed]
- Smolenski, R.T.; Simmonds, H.A.; Chambers, D.J. Exogenous Adenosine, Supplied Transiently during Reperfusion, Ameliorates Depressed Endogenous Adenosine Production in the Post-Ischemic Rat Heart. J. Mol. Cell Cardiol. 1997, 29, 333–346. [Google Scholar] [CrossRef]
- Seligmann, C.; Kupatt, C.; Becker, B.F.; Zahler, S.; Beblo, S. Adenosine Endogenously Released during Early Reperfusion Mitigates Postischemic Myocardial Dysfunction by Inhibiting Platelet Adhesion. J. Cardiovasc. Pharmacol. 1998, 32, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S. Energy Metabolism in Heart Failure and Remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Vannoni, D.; Bernini, A.; Carlucci, F.; Civitelli, S.; Di Pietro, M.C.; Leoncini, R.; Rosi, F.; Tabucchi, A.; Tanzini, G.; Marinello, E. Enzyme Activities Controlling Adenosine Levels in Normal and Neoplastic Tissues. Med. Oncol. 2004, 21, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Deussen, A. Metabolic Flux Rates of Adenosine in the Heart. Naunyn. Schmiedebergs Arch. Pharmacol. 2000, 362, 351–363. [Google Scholar] [CrossRef]
- Deussen, A.; Moser, G.; Schrader, J. Contribution of Coronary Endothelial Cells to Cardiac Adenosine Production. Pflug. Arch. 1986, 406, 608–614. [Google Scholar] [CrossRef]
- Schrader, J. Metabolism of Adenosine and Sites of Production in the Heart. In Regulatory Function of Adenosine; Springer: Boston, MA, USA, 1983; pp. 133–156. [Google Scholar]
- Newby, A.C. The Pigeon Heart 5′-Nucleotidase Responsible for Ischaemia-Induced Adenosine Formation. Biochem. J. 1988, 253, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Davila, A.; Tian, Y.; Czikora, I.; Li, J.; Su, H.; Huo, Y.; Patel, V.; Robinson, V.; Kapuku, G.; Weintraub, N.; et al. Adenosine Kinase Inhibition Augments Conducted Vasodilation and Prevents Left Ventricle Diastolic Dysfunction in Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2019, 12, e005762. [Google Scholar] [CrossRef]
- Eckle, T.; Krahn, T.; Grenz, A.; Köhler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K.; et al. Cardioprotection by Ecto-5-Nucleotidase (CD73) and A 2B Adenosine Receptors. Circulation 2007, 115, 1581–1590. [Google Scholar] [CrossRef] [Green Version]
- Osanami, A.; Sato, T.; Toda, Y.; Shimizu, M.; Kuno, A.; Kouzu, H.; Yano, T.; Ohwada, W.; Ogawa, T.; Miura, T.; et al. Adenosine Monophosphate Deaminase in the Endoplasmic Reticulum–Mitochondria Interface Promotes Mitochondrial Ca2+ Overload in Type 2 Diabetes Rat Hearts. J. Diabetes Investig. 2023. [Google Scholar] [CrossRef] [PubMed]
- Gross, M. Molecular Biology of AMP Deaminase Deficiency. Pharm. World Sci. 1994, 16, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kolek, M.J.; Carlquist, J.F.; Thaneemit-Chen, S.; Lazzeroni, L.C.; Whiting, B.M.; Horne, B.D.; Muhlestein, J.B.; Lavori, P.; Anderson, J.L. The Role of a Common Adenosine Monophosphate Deaminase (AMPD)-1 Polymorphism in Outcomes of Ischemic and Nonischemic Heart Failure. J. Card. Fail. 2005, 11, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Plaideau, C.; Lai, Y.C.; Kviklyte, S.; Zanou, N.; Lofgren, L.; Andersen, H.; Vertommen, D.; Gailly, P.; Hue, L.; Bohlooly, Y.M.; et al. Effects of Pharmacological AMP Deaminase Inhibition and Ampd1 Deletion on Nucleotide Levels and AMPK Activation in Contracting Skeletal Muscle. Chem. Biol. 2014, 21, 1497–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.T.; Dyck, J.R.B. Is AMPK the Savior of the Failing Heart? Trends Endocrinol. Metab. 2015, 26, 40–48. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: Positive and Negative Regulation, and Its Role in Whole-Body Energy Homeostasis. Curr. Opin. Cell Biol. 2014, 33c, 1–7. [Google Scholar] [CrossRef]
- Smolenski, R.T.; Jayakumar, J.; Seymour, A.-M.L.; Yacoub, M.H. The Effects of Nucleotide Pool on Purine Production in the Postischemic Heart; Springer: Boston, MA, USA, 1998; pp. 271–274. [Google Scholar]
- Jayakumar, J.; Smolenski, R.T.; Gray, C.C.; Goodwin, A.T.; Amrani, M.; Yacoub, M.H. Effects of Heat Stress on Metabolism of High-Energy Phosphates. Comparison of Normothermic and Hypothermic Ischemia. J. Cardiovasc. Surg. 1999, 40, 481–486. [Google Scholar]
- Smolenski, R.T.; Raisky, O.; Slominska, E.M.; Abunasra, H.; Kalsi, K.K.; Jayakumar, J.; Suzuki, K.; Yacoub, M.H. Protection from Reperfusion Injury after Cardiac Transplantation by Inhibition of Adenosine Metabolism and Nucleotide Precursor Supply. Circulation 2001, 104, I246–I252. [Google Scholar] [CrossRef] [Green Version]
- Zaha, V.G.; Young, L.H. AMP-Activated Protein Kinase Regulation and Biological Actions in the Heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef] [Green Version]
- Perings, S.M.; Schulze, K.; Decking, U.; Kelm, M.; Strauer, B.E. Age-Related Decline of PCr/ATP-Ratio in Progressively Hypertrophied Hearts of Spontaneously Hypertensive Rats. Heart Vessel. 2000, 15, 197–202. [Google Scholar] [CrossRef]
- Gustafson, L.A.; Kroll, K. Downregulation of 5′-Nucleotidase in Rabbit Heart during Coronary Underperfusion. Am. J. Physiol. Circ. Physiol. 1998, 274, H529–H538. [Google Scholar] [CrossRef] [PubMed]
- Le, G.Y.; Essackjee, H.C.; Ballard, H.J. Intracellular Adenosine Formation and Release by Freshly-Isolated Vascular Endothelial Cells from Rat Skeletal Muscle: Effects of Hypoxia and/or Acidosis. Biochem. Biophys. Res. Commun. 2014, 450, 93–98. [Google Scholar] [CrossRef]
- Bak, M.I.; Ingwall, J.S. Acidosis during Ischemia Promotes Adenosine Triphosphate Resynthesis in Postischemic Rat Heart. In Vivo Regulation of 5’-Nucleotidase. J. Clin. Investig. 1994, 93, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia: A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, H.; Rauen, U. Ischemia-Reperfusion Injury: Processes in Pathogenetic Networks: A Review. Transplant. Proc. 2007, 39, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Katayama, O.; Ledingham, S.J.; Amrani, M.; Smolenski, R.T.; Lachno, D.R.; Jayakumar, J.; Yacoub, M.H. Functional and Metabolic Effects of Adenosine in Cardioplegia: Role of Temperature and Concentration. Ann. Thorac. Surg. 1997, 63, 445–449. [Google Scholar] [CrossRef]
- Savabi, F. Free Creatine Available to the Creatine Phosphate Energy Shuttle in Isolated Rat Atria. Proc. Natl. Acad. Sci. USA 1988, 85, 7476–7480. [Google Scholar] [CrossRef] [Green Version]
- Semenovsky, M.L.; Shumakov, V.I.; Sharov, V.G.; Mogilevsky, G.M.; Asmolovsky, A.V.; Makhotina, L.A.; Saks, V.A. Protection of Ischemic Myocardium by Exogenous Phosphocreatine. II. Clinical, Ultrastructural, and Biochemical Evaluations. J. Thorac. Cardiovasc. Surg. 1987, 94, 762–769. [Google Scholar] [CrossRef]
- Smolenski, R.T.; Yacoub, M.H.; Seymour, A.-M.L. Reduced Purine Catabolite Production in the Postischemic Rat Heart: A31P NMR Assessment of Cytosolic Metabolites. Magma Magn. Reson. Mater. Phys. Biol. Med. 1994, 2, 417–420. [Google Scholar] [CrossRef]
- Reimer, K.A.; Murry, C.E.; Yamasawa, I.; Hill, M.L.; Jennings, R.B. Four Brief Periods of Myocardial Ischemia Cause No Cumulative ATP Loss or Necrosis. Am. J. Physiol. 1986, 251, H1306–H1315. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Roberts, A.F.; Raine, A.E. Energy Metabolism in Reperfused Heart Muscle: Metabolic Correlates to Return of Function. J. Am. Coll. Cardiol. 1985, 6, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BUNGER, R.; SOBOLL, S. Cytosolic Adenylates and Adenosine Release in Perfused Working Heart. Comparison of Whole Tissue with Cytosolic Non-Aqueous Fractionation Analyses. Eur. J. Biochem. 1986, 159, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy Substrate Metabolism and Mitochondrial Oxidative Stress in Cardiac Ischemia/Reperfusion Injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Shneyvays, V.; Leshem, D.; Zinman, T.; Mamedova, L.K.; Jacobson, K.A.; Shainberg, A. Role of Adenosine A1 and A3 Receptors in Regulation of Cardiomyocyte Homeostasis after Mitochondrial Respiratory Chain Injury. Am. J. Physiol. -Hear. Circ. Physiol. 2005, 288, H2792–H2801. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Pelleg, A. Cardiac Purinergic Signalling in Health and Disease. Purinergic Signal. 2015, 11, 1–46. [Google Scholar] [CrossRef]
- Neely, J.; Liebermeister, H.; Battersby, E.; Morgan, H. Effect of Pressure Development on Oxygen Consumption by Isolated Rat Heart. Am. J. Physiol. Content 1967, 212, 804–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle Isolation: Functional Mitochondria from Mouse Liver, Muscle and Cultured Filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Smolenski, R.T.; Lachno, D.R.; Ledingham, S.J.M.; Yacoub, M.H. Determination of 16 Nucleotides, Nucleosides and Bases Using High-Performance Liquid-Chromatography and Its Application to the Study of Purine Metabolism in Hearts for Transplantation. J. Chromatogr. Appl. 1990, 527, 414–420. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Seymour, A.-M.L. Imaging Cardiac Metabolism in Heart Failure: The Potential of NMR Spectroscopy in the Era of Metabolism Revisited. Hear. Lung Circ. 2003, 12, 25–30. [Google Scholar] [CrossRef]
- Roth, K.; Weiner, M.W. Determination of Cytosolic ADP and AMP Concentrations and the Free Energy of ATP Hydrolysis in Human Muscle and Brain Tissues with 31P NMR Spectroscopy. Magn. Reson. Med. 1991, 22, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Braczko, A.; Kutryb-Zajac, B.; Jedrzejewska, A.; Krol, O.; Mierzejewska, P.; Zabielska-Kaczorowska, M.; Slominska, E.M.; Smolenski, R.T. Cardiac Mitochondria Dysfunction in Dyslipidemic Mice. Int. J. Mol. Sci. 2022, 23, 11488. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zabielska-Kaczorowska, M.A.; Braczko, A.; Pelikant-Malecka, I.; Slominska, E.M.; Smolenski, R.T. Hidden Pool of Cardiac Adenine Nucleotides That Controls Adenosine Production. Pharmaceuticals 2023, 16, 599. https://doi.org/10.3390/ph16040599
Zabielska-Kaczorowska MA, Braczko A, Pelikant-Malecka I, Slominska EM, Smolenski RT. Hidden Pool of Cardiac Adenine Nucleotides That Controls Adenosine Production. Pharmaceuticals. 2023; 16(4):599. https://doi.org/10.3390/ph16040599
Chicago/Turabian StyleZabielska-Kaczorowska, Magdalena A., Alicja Braczko, Iwona Pelikant-Malecka, Ewa M. Slominska, and Ryszard T. Smolenski. 2023. "Hidden Pool of Cardiac Adenine Nucleotides That Controls Adenosine Production" Pharmaceuticals 16, no. 4: 599. https://doi.org/10.3390/ph16040599