
Glycosylation and Lipidation Strategies: Approaches for Improving Antimicrobial Peptide Efficacy


Abstract
:1. Introduction
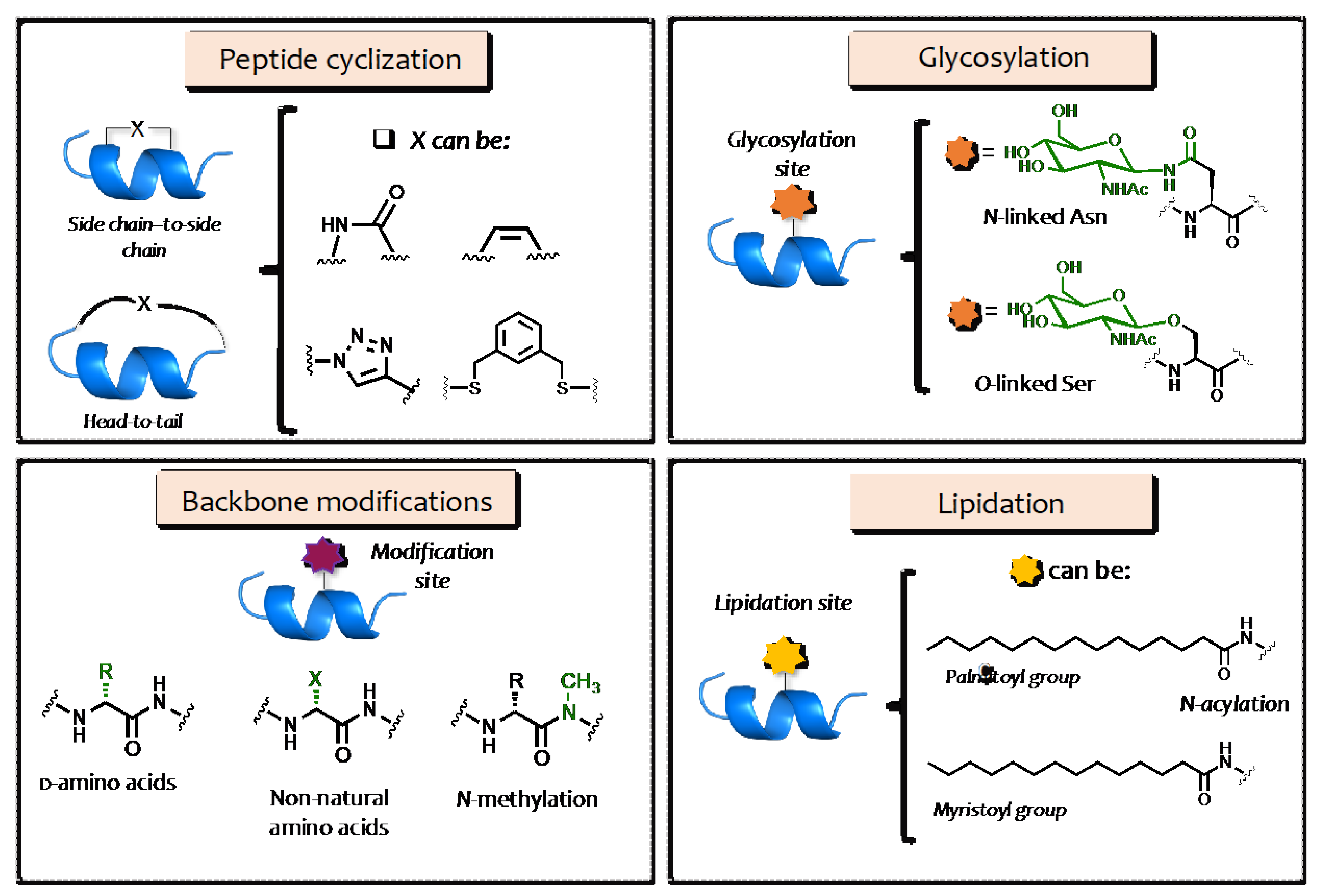
2. Structural Modification of AMPs
3. AMP Glycosylation
3.1. O–Glycosylation
3.2. N–Glycosilation
3.3. S–Glycosilation
3.4. C–Glycosylation
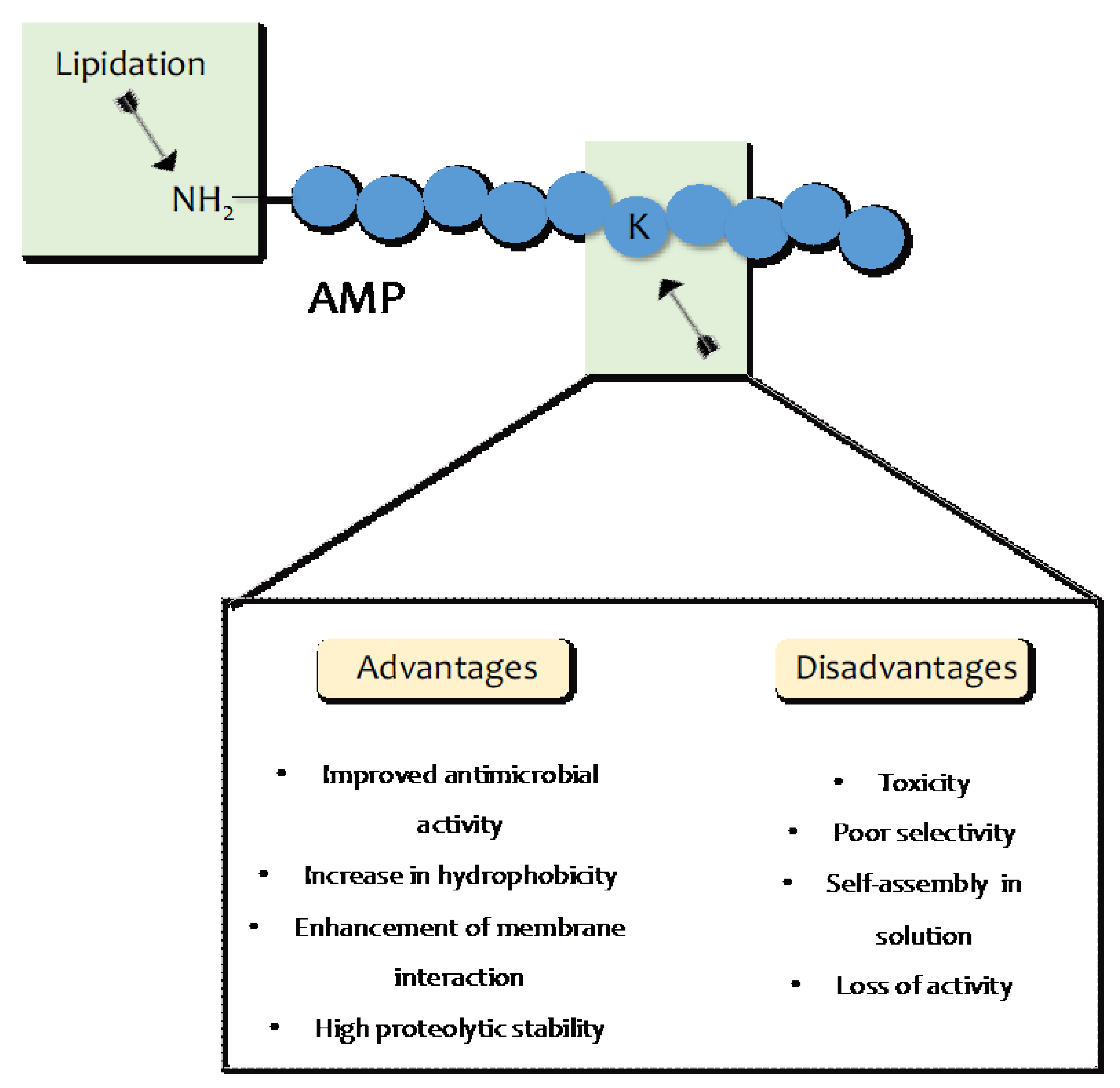
4. AMP Lipidation
5. Applications of Glycosylation and Lipidation to Improve Activity through Enhancing Delivery
6. Conclusions Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A Review on Antibiotic Resistance: Alarm Bells are Ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef] [Green Version]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [Green Version]
- Brogden, K. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef]
- Falanga, A.; Maione, A.; La Pietra, A.; de Alteriis, E.; Vitale, S.; Bellavita, R.; Carotenuto, R.; Turra, D.; Galdiero, S.; Galdiero, E.; et al. Competitiveness during Dual-Species Biofilm Formation of Fusarium oxysporum and Candida albicans and a Novel Treatment Strategy. Pharmaceutics 2022, 14, 1167. [Google Scholar] [CrossRef]
- Maione, A.; Bellavita, R.; de Alteriis, E.; Galdiero, S.; Albarano, L.; La Pietra, A.; Guida, M.; Parrilli, E.; D’Angelo, C.; Galdiero, E.; et al. WMR Peptide as Antifungal and Antibiofilm against Albicans and Non-Albicans Candida Species: Shreds of Evidence on the Mechanism of Action. Int. J. Mol. Sci. 2022, 23, 2151. [Google Scholar] [CrossRef]
- Dini, I.; De Biasi, M.G.; Mancusi, A. An Overview of the Potentialities of Antimicrobial Peptides Derived from Natural Sources. Antibiotics 2022, 11, 1483. [Google Scholar] [CrossRef]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.D.; Lin, F.; Hossain, M.A.; Dawson, R.M. Chemical synthesis and biological evaluation of an antimicrobial peptide gonococcal growth inhibitor. Amino Acids 2012, 43, 2279–2283. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.X.L.; Dong, C. Antimicrobial Peptides: An Overview of their Structure, Function and Mechanism of Action. Protein Pept. Lett. 2022, 29, 641–650. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2580. [Google Scholar] [CrossRef] [PubMed]
- Bellavita, R.; Raucci, F.; Merlino, F.; Piccolo, M.; Ferraro, M.G.; Irace, C.; Santamaria, R.; Iqbal, A.J.; Novellino, E.; Grieco, P.; et al. Temporin L-derived peptide as a regulator of the acute inflammatory response in zymosan-induced peritonitis. Biomed. Pharmacother. 2020, 123, 109788. [Google Scholar] [CrossRef]
- Powers, J.P.S.; Hancock, R.E.W. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef]
- Bellavita, R.; Buommino, E.; Casciaro, B.; Merlino, F.; Cappiello, F.; Marigliano, N.; Saviano, A.; Maione, F.; Santangelo, R.; Mangoni, M.L.; et al. Synthetic Amphipathic beta-Sheet Temporin-Derived Peptide with Dual Antibacterial and Anti-Inflammatory Activities. Antibiotics 2022, 11, 1285. [Google Scholar] [CrossRef]
- Cheng, Q.; Zeng, P. Hydrophobic-hydrophilic Alternation: An effective Pattern to de novo Designed Antimicrobial Peptides. Curr. Pharm. Des. 2022, 28, 3527–3537. [Google Scholar]
- Bradshaw, J. Cationic antimicrobial peptides: Issues for potential clinical use. BioDrugs 2003, 17, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides With Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramazi, S.; Mohammadi, N.; Allahverdi, A.; Khalili, E.; Abdolmaleki, P. A review on antimicrobial peptides databases and the computational tools. Database 2022, 2022, baac011. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. BBA Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [Green Version]
- Sani, M.A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef]
- Bellavita, R.; Vollaro, A.; Catania, M.R.; Merlino, F.; De Martino, L.; Nocera, F.P.; DellaGreca, M.; Lembo, F.; Grieco, P.; Buommino, E. Novel Antimicrobial Peptide from Temporin L in The Treatment of Staphylococcus pseudintermedius and Malassezia pachydermatis in Polymicrobial Inter-Kingdom Infection. Antibiotics 2020, 9, 530. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Buommino, E.; Carotenuto, A.; Antignano, I.; Bellavita, R.; Casciaro, B.; Loffredo, M.R.; Merlino, F.; Novellino, E.; Mangoni, M.L.; Nocera, F.P.; et al. The Outcomes of Decorated Prolines in the Discovery of Antimicrobial Peptides from Temporin-L. ChemMedChem 2019, 14, 1283–1290. [Google Scholar] [CrossRef]
- Yousif, A.M.; Ingangi, V.; Merlino, F.; Brancaccio, D.; Minopoli, M.; Bellavita, R.; Novellino, E.; Carriero, M.V.; Carotenuto, A.; Grieco, P. Urokinase receptor derived peptides as potent inhibitors of the formyl peptide receptor type 1-triggered cell migration. Eur. J. Med. Chem. 2018, 143, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Bellavita, R.; Maione, A.; Merlino, F.; Siciliano, A.; Dardano, P.; De Stefano, L.; Galdiero, S.; Galdiero, E.; Grieco, P.; Falanga, A. Antifungal and Antibiofilm Activity of Cyclic Temporin L Peptide Analogues against Albicans and Non-Albicans Candida Species. Pharmaceutics 2022, 14, 454. [Google Scholar] [CrossRef] [PubMed]
- Bellavita, R.; Casciaro, B.; Di Maro, S.; Brancaccio, D.; Carotenuto, A.; Falanga, A.; Cappiello, F.; Buommino, E.; Galdiero, S.; Novellino, E.; et al. First-in-Class Cyclic Temporin L Analogue: Design, Synthesis, and Antimicrobial Assessment. J. Med. Chem. 2021, 64, 11675–11694. [Google Scholar] [CrossRef]
- Wendt, M.; Bellavita, R.; Gerber, A.; Efrem, N.L.; van Ramshorst, T.; Pearce, N.M.; Davey, P.R.J.; Everard, I.; Vazquez-Chantada, M.; Chiarparin, E.; et al. Bicyclic beta-Sheet Mimetics that Target the Transcriptional Coactivator beta-Catenin and Inhibit Wnt Signaling. Angew. Chem. Int. Ed. Engl. 2021, 60, 13937–13944. [Google Scholar] [CrossRef] [PubMed]
- Merlino, F.; Billard, E.; Yousif, A.M.; Di Maro, S.; Brancaccio, D.; Abate, L.; Carotenuto, A.; Bellavita, R.; Bianca, R.D.D.V.; Santicioli, P.; et al. Functional Selectivity Revealed by N-Methylation Scanning of Human Urotensin II and Related Peptides. J. Med. Chem. 2019, 62, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Oren, Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides 2001, 22, 1629–1641. [Google Scholar] [CrossRef]
- Scudiero, O.; Nigro, E.; Cantisani, M.; Colavita, I.; Leone, M.; Mercurio, F.A.; Galdiero, M.; Pessi, A.; Daniele, A.; Salvatore, F.; et al. Design and activity of a cyclic mini-beta-defensin analog: A novel antimicrobial tool. Int. J. Nanomed. 2015, 10, 6523–6539. [Google Scholar]
- Manteghi, R.; Pallagi, E.; Olajos, G.; Csoka, I. Pegylation and formulation strategy of Anti-Microbial Peptide (AMP) according to the quality by design approach. Eur. J. Pharm. Sci. 2020, 144, 105197. [Google Scholar] [CrossRef]
- Brito, J.C.M.; Carvalho, L.R.; Neves de Souza, A.; Carneiro, G.; Magalhaes, P.P.; Farias, L.M.; Guimaraes, N.R.; Verly, R.M.; Resende, J.M.; Elena de Lima, M. PEGylation of the antimicrobial peptide LyeTx I-b maintains structure-related biological properties and improves selectivity. Front. Mol. Biosci. 2022, 9, 1001508. [Google Scholar] [CrossRef]
- Hamley, I.W. PEG-peptide conjugates. Biomacromolecules 2014, 15, 1543–1559. [Google Scholar] [CrossRef] [PubMed]
- Bednarska, N.G.; Wren, B.W.; Willcocks, S.J. The importance of the glycosylation of antimicrobial peptides: Natural and synthetic approaches. Drug Discov. Today 2017, 22, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zhang, X.; Chen, X.; Aramsangtienchai, P.; Tong, Z.; Lin, H. Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev. 2018, 118, 919–988. [Google Scholar] [CrossRef] [PubMed]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef]
- Moradi, S.V.; Hussein, W.M.; Varamini, P.; Simerska, P.; Toth, I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7, 2492–2500. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; Nelson, C.F.; Nusslein, K.; Tew, G.N. Hydrophilic modifications of an amphiphilic polynorbornene and the effects on its hemolytic and antibacterial activity. Biomacromolecules 2009, 10, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef]
- Wang, G. Post-translational Modifications of Natural Antimicrobial Peptides and Strategies for Peptide Engineering. Curr. Biotechnol. 2012, 1, 72–79. [Google Scholar] [CrossRef]
- Wormald, M.R.; Wooten, E.W.; Bazzo, R.; Edge, C.J.; Feinstein, A.; Rademacher, T.W.; Dwek, R.A. The conformational effects of N-glycosylation on the tailpiece from serum IgM. Eur. J. Biochem. 1991, 198, 131–139. [Google Scholar] [CrossRef]
- Lommerse, J.P.; Kroon-Batenburg, L.M.; Kroon, J.; Kamerling, J.P.; Vliegenthart, J.F. Conformations and internal mobility of a glycopeptide derived from bromelain using molecular dynamics simulations and NOESY analysis. J. Biomol. NMR 1995, 6, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Bosques, C.J.; Imperiali, B. The interplay of glycosylation and disulfide formation influences fibrillization in a prion protein fragment. Proc. Natl. Acad. Sci. USA 2003, 100, 7593–7598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Conner, S.E.; Imperiali, B. A molecular basis for glycosylation-induced conformational switching. Chem Biol 1998, 5, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Chang, X.; Wang, Y.; Gao, G.F.; Shao, Y.; Ma, L.; Li, X. Glycosylated enfuvirtide: A long-lasting glycopeptide with potent anti-HIV activity. J. Med. Chem. 2015, 58, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Tarp, M.A.; Clausen, H. Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim. Biophys. Acta 2008, 1780, 546–563. [Google Scholar] [CrossRef]
- Lele, D.S.; Talat, S.; Kumari, S.; Srivastava, N.; Kaur, K.J. Understanding the importance of glycosylated threonine and stereospecific action of Drosocin, a Proline rich antimicrobial peptide. Eur. J. Med. Chem. 2015, 92, 637–647. [Google Scholar] [CrossRef]
- Talat, S.; Thiruvikraman, M.; Kumari, S.; Kaur, K.J. Glycosylated analogs of formaecin I and drosocin exhibit differential pattern of antibacterial activity. Glycoconj. J. 2011, 28, 537–555. [Google Scholar] [CrossRef]
- Bulet, P.; Dimarcq, J.L.; Hetru, C.; Lagueux, M.; Charlet, M.; Hegy, G.; Van Dorsselaer, A.; Hoffmann, J.A. A novel inducible antibacterial peptide of Drosophila carries an O-glycosylated substitution. J. Biol. Chem. 1993, 268, 14893–14897. [Google Scholar] [CrossRef] [PubMed]
- McManus, A.M.; Otvos, L., Jr.; Hoffmann, R.; Craik, D.J. Conformational studies by NMR of the antimicrobial peptide, drosocin, and its non-glycosylated derivative: Effects of glycosylation on solution conformation. Biochemistry 1999, 38, 705–714. [Google Scholar] [CrossRef]
- Bulet, P.; Urge, L.; Ohresser, S.; Hetru, C.; Otvos, L., Jr. Enlarged scale chemical synthesis and range of activity of drosocin, an O-glycosylated antibacterial peptide of Drosophila. Eur. J. Biochem. 1996, 238, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Coltart, D.M.; Royyuru, A.K.; Williams, L.J.; Glunz, P.W.; Sames, D.; Kuduk, S.D.; Schwarz, J.B.; Chen, X.T.; Danishefsky, S.J.; Live, D.H. Principles of mucin architecture: Structural studies on synthetic glycopeptides bearing clustered mono-, di-, tri-, and hexasaccharide glycodomains. J. Am. Chem. Soc. 2002, 124, 9833–9844. [Google Scholar] [CrossRef]
- Lele, D.S.; Dwivedi, R.; Kumari, S.; Kaur, K.J. Effect of distal sugar and interglycosidic linkage of disaccharides on the activity of proline rich antimicrobial glycopeptides. J. Pept. Sci. 2015, 21, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, J.A.; Veal, D.A.; Beattie, A.J.; Gooley, A.A. Isolation from an ant Myrmecia gulosa of two inducible O-glycosylated proline-rich antibacterial peptides. J. Biol. Chem. 1998, 273, 6139–6143. [Google Scholar] [CrossRef] [Green Version]
- Nesuta, O.; Hexnerova, R.; Budesinsky, M.; Slaninova, J.; Bednarova, L.; Hadravova, R.; Straka, J.; Veverka, V.; Cerovsky, V. Antimicrobial Peptide from the Wild Bee Hylaeus signatus Venom and Its Analogues: Structure-Activity Study and Synergistic Effect with Antibiotics. J. Nat. Prod. 2016, 79, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Ai, S.; Chen, Q.; Chen, X.Y.; Li, H.J.; Li, Y.L.; Zhao, X. Effects of Glycosylation and d-Amino Acid Substitution on the Antitumor and Antibacterial Activities of Bee Venom Peptide HYL. Bioconjugate Chem. 2020, 31, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Maky, M.A.; Ishibashi, N.; Zendo, T.; Perez, R.H.; Doud, J.R.; Karmi, M.; Sonomoto, K. Enterocin F4-9, a Novel O-Linked Glycosylated Bacteriocin. Appl. Environ. Microbiol. 2015, 81, 4819–4826. [Google Scholar] [CrossRef] [Green Version]
- Humisto, A.; Jokela, J.; Teigen, K.; Wahlsten, M.; Permi, P.; Sivonen, K.; Herfindal, L. Characterization of the interaction of the antifungal and cytotoxic cyclic glycolipopeptide hassallidin with sterol-containing lipid membranes. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1510–1521. [Google Scholar] [CrossRef]
- Vestola, J.; Shishido, T.K.; Jokela, J.; Fewer, D.P.; Aitio, O.; Permi, P.; Wahlsten, M.; Wang, H.; Rouhiainen, L.; Sivonen, K. Hassallidins, antifungal glycolipopeptides, are widespread among cyanobacteria and are the end-product of a nonribosomal pathway. Proc. Natl. Acad. Sci. USA 2014, 111, E1909–E1917. [Google Scholar] [CrossRef] [Green Version]
- Egleton, R.D.; Mitchell, S.A.; Huber, J.D.; Palian, M.M.; Polt, R.; Davis, T.P. Improved blood-brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J. Pharmacol. Exp. Ther. 2001, 299, 967–972. [Google Scholar]
- Mellquist, J.L.; Kasturi, L.; Spitalnik, S.L.; Shakin-Eshleman, S.H. The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 1998, 37, 6833–6837. [Google Scholar] [CrossRef]
- Shakin-Eshleman, S.H.; Spitalnik, S.L.; Kasturi, L. The amino acid at the X position of an Asn-X-Ser sequon is an important determinant of N-linked core-glycosylation efficiency. J. Biol. Chem. 1996, 271, 6363–6366. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Xue, J.; Swarts, B.M.; Wang, Q.; Wu, Q.; Guo, Z. Synthesis and antibacterial activities of N-glycosylated derivatives of tyrocidine A, a macrocyclic peptide antibiotic. J. Med. Chem. 2009, 52, 2052–2059. [Google Scholar] [CrossRef]
- Zou, Y.; Zhao, Q.; Zhang, C.; Wang, L.; Li, W.; Li, X.; Wu, Q.; Hu, H. Synthesis and antibacterial activities of novel tyrocidine A glycosylated derivatives towards multidrug-resistant pathogens. J. Pept. Sci. 2015, 21, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M. A novel hydroxyproline rich glycopeptide from pericarp of Datura stramonium: Proficiently eradicate the biofilm of antifungals resistant Candida albicans. Biopolymers 2012, 98, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, E.; Collis, D.W.P.; Mikut, R.; Hilpert, K. The effect of lipidation and glycosylation on short cationic antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183195. [Google Scholar] [CrossRef]
- Stepper, J.; Shastri, S.; Loo, T.S.; Preston, J.C.; Novak, P.; Man, P.; Moore, C.H.; Havlicek, V.; Patchett, M.L.; Norris, G.E. Cysteine S-glycosylation, a new post-translational modification found in glycopeptide bacteriocins. FEBS Lett. 2011, 585, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amso, Z.; Bisset, S.W.; Yang, S.H.; Harris, P.W.R.; Wright, T.H.; Navo, C.D.; Patchett, M.L.; Norris, G.E.; Brimble, M.A. Total chemical synthesis of glycocin F and analogues: S-glycosylation confers improved antimicrobial activity. Chem. Sci. 2018, 9, 1686–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oman, T.J.; Boettcher, J.M.; Wang, H.; Okalibe, X.N.; van der Donk, W.A. Sublancin is not a lantibiotic but an S-linked glycopeptide. Nat. Chem. Biol. 2011, 7, 78–80. [Google Scholar] [CrossRef]
- Hsieh, Y.S.; Wilkinson, B.L.; O’Connell, M.R.; Mackay, J.P.; Matthews, J.M.; Payne, R.J. Synthesis of the bacteriocin glycopeptide sublancin 168 and S-glycosylated variants. Org. Lett. 2012, 14, 1910–1913. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- El Malah, T.; Nour, H.F.; Satti, A.A.E.; Hemdan, B.A.; El-Sayed, W.A. Design, Synthesis, and Antimicrobial Activities of 1,2,3-Triazole Glycoside Clickamers. Molecules 2020, 25, 790. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Xia, Y.N.; Gulzar, T.; Wei, B.; Li, H.; Zhu, D.; Hu, Z.; Xu, P.; Yu, B. Facile access to C-glycosyl amino acids and peptides via Ni-catalyzed reductive hydroglycosylation of alkynes. Nat. Commun. 2021, 12, 4924. [Google Scholar] [CrossRef]
- Junior, E.F.C.; Guimaraes, C.; Franco, L.L.; Alves, R.J.; Kato, K.C.; Martins, H.R.; Filho, J.D.d.S.; Bemquerer, M.P.; Munhoz, V.H.O.; Resende, J.M.; et al. Glycotriazole-peptides derived from the peptide HSP1: Synergistic effect of triazole and saccharide rings on the antifungal activity. Amino Acids 2017, 49, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Groothuys, S.; Heredia, A.; Kuijpers, B.H.; Rutjes, F.P.; van Delft, F.L.; Wang, L.X. Enzymatic glycosylation of triazole-linked GlcNAc/Glc-peptides: Synthesis, stability and anti-HIV activity of triazole-linked HIV-1 gp41 glycopeptide C34 analogues. ChemBioChem 2009, 10, 1234–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rounds, T.; Straus, S.K. Lipidation of Antimicrobial Peptides as a Design Strategy for Future Alternatives to Antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef] [PubMed]
- Rezende, S.B.; Oshiro, K.G.N.; Junior, N.G.O.; Franco, O.L.; Cardoso, M.H. Advances on chemically modified antimicrobial peptides for generating peptide antibiotics. Chem. Commun. 2021, 57, 11578–11590. [Google Scholar] [CrossRef] [PubMed]
- Kamysz, E.; Sikorska, E.; Jaskiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Baranska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12-Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef] [Green Version]
- Bellavita, R.; Falanga, A.; Buommino, E.; Merlino, F.; Casciaro, B.; Cappiello, F.; Mangoni, M.L.; Novellino, E.; Catania, M.R.; Paolillo, R.; et al. Novel temporin L antimicrobial peptides: Promoting self-assembling by lipidic tags to tackle superbugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1751–1764. [Google Scholar] [CrossRef]
- Malina, A.; Shai, Y. Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem. J. 2005, 390, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Humpola, M.V.; Rey, M.C.; Carballeira, N.M.; Simonetta, A.C.; Tonarelli, G.G. Biological and structural effects of the conjugation of an antimicrobial decapeptide with saturated, unsaturated, methoxylated and branched fatty acids. J. Pept. Sci. 2017, 23, 45–55. [Google Scholar] [CrossRef]
- Bellavita, R.; Annarita, F.; Merlino, F.; D’Auria, G.; Molfetta, N.; Saviano, A.; Maione, F.; Galdiero, U.; Catania, M.R.; Stefania, G.; et al. Unveiling the mechanism of action of acylated temporin L analogues against multidrug-resistant Candida albicans. J. Enzym. Inhib. Med. Chem. 2023, 38, 36–50. [Google Scholar] [CrossRef]
- Roscetto, E.; Bellavita, R.; Paolillo, R.; Merlino, F.; Molfetta, N.; Grieco, P.; Buommino, E.; Catania, M.R. Antimicrobial Activity of a Lipidated Temporin L Analogue against Carbapenemase-Producing Klebsiella pneumoniae Clinical Isolates. Antibiotics 2021, 1, 1312. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.; Pohl, J.; Dubin, A.; Reed, M.S.; Bowers, S.E.; Fallon, M.T.; Shafer, W.M. The increased bactericidal activity of a fatty acid-modified synthetic antimicrobial peptide of human cathepsin G correlates with its enhanced capacity to interact with model membranes. Int. J. Antimicrob. Agents 2003, 21, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohan, S.; Cameotra, S.S.; Bisht, G.S. Systematic study of non-natural short cationic lipopeptides as novel broad-spectrum antimicrobial agents. Chem. Biol. Drug Des. 2013, 82, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Lohan, S.; Monga, J.; Cameotra, S.S.; Bisht, G.S. In vitro and in vivo antibacterial evaluation and mechanistic study of ornithine based small cationic lipopeptides against antibiotic resistant clinical isolates. Eur. J. Med. Chem. 2014, 88, 19–27. [Google Scholar] [CrossRef]
- Sikora, K.; Bauer, M.; Bartoszewska, S.; Neubauer, D.; Kamysz, W. Glycosylated Lipopeptides-Synthesis and Evaluation of Antimicrobial Activity and Cytotoxicity. Biomolecules 2023, 13, 172. [Google Scholar] [CrossRef]
- Jensen, S.K.; Thomsen, T.T.; Oddo, A.; Franzyk, H.; Lobner-Olesen, A.; Hansen, P.R. Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position. Int. J. Mol. Sci. 2020, 21, 5829. [Google Scholar] [CrossRef]
- Liao, G.; Shi, T.; Xie, J. Regulation mechanisms underlying the biosynthesis of daptomycin and related lipopeptides. J. Cell Biochem. 2012, 113, 735–741. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Hood, K.S.; Mileykovskaya, E.; Miller, W.R.; Tran, T.T. Bacterial cell membranes and their role in daptomycin resistance: A review. Front. Mol. Biosci. 2022, 9, 1035574. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, S.S.; Baindara, P.; Sharma, S.; Khatri, N.; Grover, V.; Patil, P.B.; Korpole, S. Surfactin Like Broad Spectrum Antimicrobial Lipopeptide Co-produced With Sublancin From Bacillus subtilis Strain A52: Dual Reservoir of Bioactives. Front. Microbiol. 2020, 11, 1167. [Google Scholar] [CrossRef]
- Carrillo, C.; Teruel, J.A.; Aranda, F.J.; Ortiz, A. Molecular mechanism of membrane permeabilization by the peptide antibiotic surfactin. Biochim. Biophys. Acta 2003, 1611, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, S.; Romo, T.D.; Grossfield, A. Selectivity and Mechanism of Fengycin, an Antimicrobial Lipopeptide, from Molecular Dynamics. J. Phys. Chem. B 2018, 122, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Vanittanakom, N.; Loeffler, W.; Koch, U.; Jung, G. Fengycin--a novel antifungal lipopeptide antibiotic produced by Bacillus subtilis F-29-3. J. Antibiot. 1986, 39, 888–901. [Google Scholar] [CrossRef] [Green Version]
- Wyche, T.P.; Hou, Y.; Vazquez-Rivera, E.; Braun, D.; Bugni, T.S. Peptidolipins B-F, antibacterial lipopeptides from an ascidian-derived Nocardia sp. J. Nat. Prod. 2012, 75, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Palomba, L.; Galdiero, M.; Franci, G.; Galdiero, S. A boost to the antiviral activity: Cholesterol tagged peptides derived from glycoprotein B of Herpes Simplex virus type I. Int. J. Biol. Macromol. 2020, 162, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Del Genio, V.; Bellavita, R.; Falanga, A.; Herve-Aubert, K.; Chourpa, I.; Galdiero, S. Peptides to Overcome the Limitations of Current Anticancer and Antimicrobial Nanotherapies. Pharmaceutics 2022, 14, 1235. [Google Scholar] [CrossRef]
- Chen, W.; Yang, S.; Li, S.; Lang, J.C.; Mao, C.; Kroll, P.; Tang, L.; Dong, H. Self-Assembled Peptide Nanofibers Display Natural Antimicrobial Peptides to Selectively Kill Bacteria without Compromising Cytocompatibility. ACS Appl. Mater. Interfaces 2019, 11, 28681–28689. [Google Scholar] [CrossRef]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; de Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Del Genio, V.; Falanga, A.; Allard-Vannier, E.; Herve-Aubert, K.; Leone, M.; Bellavita, R.; Uzbekov, R.; Chourpa, I.; Galdiero, S. Design and Validation of Nanofibers Made of Self-Assembled Peptides to Become Multifunctional Stimuli-Sensitive Nanovectors of Anticancer Drug Doxorubicin. Pharmaceutics 2022, 14, 1544. [Google Scholar] [CrossRef]
- Sahariah, P.; Sorensen, K.K.; Hjalmarsdottir, M.A.; Sigurjonsson, O.E.; Jensen, K.J.; Masson, M.; Thygesen, M.B. Antimicrobial peptide shows enhanced activity and reduced toxicity upon grafting to chitosan polymers. Chem. Commun. 2015, 51, 11611–11614. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.M.; Maisetta, G.; Sandreschi, S.; Gazzarri, M.; Bartoli, C.; Grassi, L.; Esin, S.; Chiellini, F.; Batoni, G. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front. Microbiol. 2015, 6, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayathilaka, E.; Nikapitiya, C.; De Zoysa, M.; Whang, I. Antimicrobial Peptide Octominin-Encapsulated Chitosan Nanoparticles Enhanced Antifungal and Antibacterial Activities. Int. J. Mol. Sci. 2022, 23, 15882. [Google Scholar] [CrossRef] [PubMed]
- Papademetriou, I.T.; Porter, T. Promising approaches to circumvent the blood-brain barrier: Progress, pitfalls and clinical prospects in brain cancer. Ther. Deliv. 2015, 6, 989–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, K.A.; Gillespie, T.J.; Huber, J.D.; Egleton, R.D.; Davis, T.P. Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides 2001, 22, 2329–2343. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipopeptide | Source | Chain Length | Activity |
|---|---|---|---|
| Daptomycin | Streptomyces roseosporus | C10 | Antibacterial |
| Surfactin | Bacillus subtilis | C16 | Antibacterial, antiviral |
| Fengycin | Bacillus subtilis | C16, | Fungicide |
| Peptidolipins B–F | Nocardia sp. | C23, C25, C27, olefin cyclopropane | Antibacterial |
| Friulimicin B | Actinoplanes friuliensis | C14 | Antibacterial |
| Arylomycin A2 | Streptomyces sp. | C12 | Antibacterial |
| Globomycin | Streptomyces hagronensis | C6 | Antibacterial |
| Tsushimycin | Streptomyces sp. | C14 | Antitrypanosomal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellavita, R.; Braccia, S.; Galdiero, S.; Falanga, A. Glycosylation and Lipidation Strategies: Approaches for Improving Antimicrobial Peptide Efficacy. Pharmaceuticals 2023, 16, 439. https://doi.org/10.3390/ph16030439
Bellavita R, Braccia S, Galdiero S, Falanga A. Glycosylation and Lipidation Strategies: Approaches for Improving Antimicrobial Peptide Efficacy. Pharmaceuticals. 2023; 16(3):439. https://doi.org/10.3390/ph16030439
Chicago/Turabian StyleBellavita, Rosa, Simone Braccia, Stefania Galdiero, and Annarita Falanga. 2023. "Glycosylation and Lipidation Strategies: Approaches for Improving Antimicrobial Peptide Efficacy" Pharmaceuticals 16, no. 3: 439. https://doi.org/10.3390/ph16030439