

Current Trends in Neoantigen-Based Cancer Vaccines
 , , , ,
, , , ,
Abstract
:1. Introduction of Fundamental Properties of T-Cell Immunity
2. Roles of Neoantigens in Immunotherapy
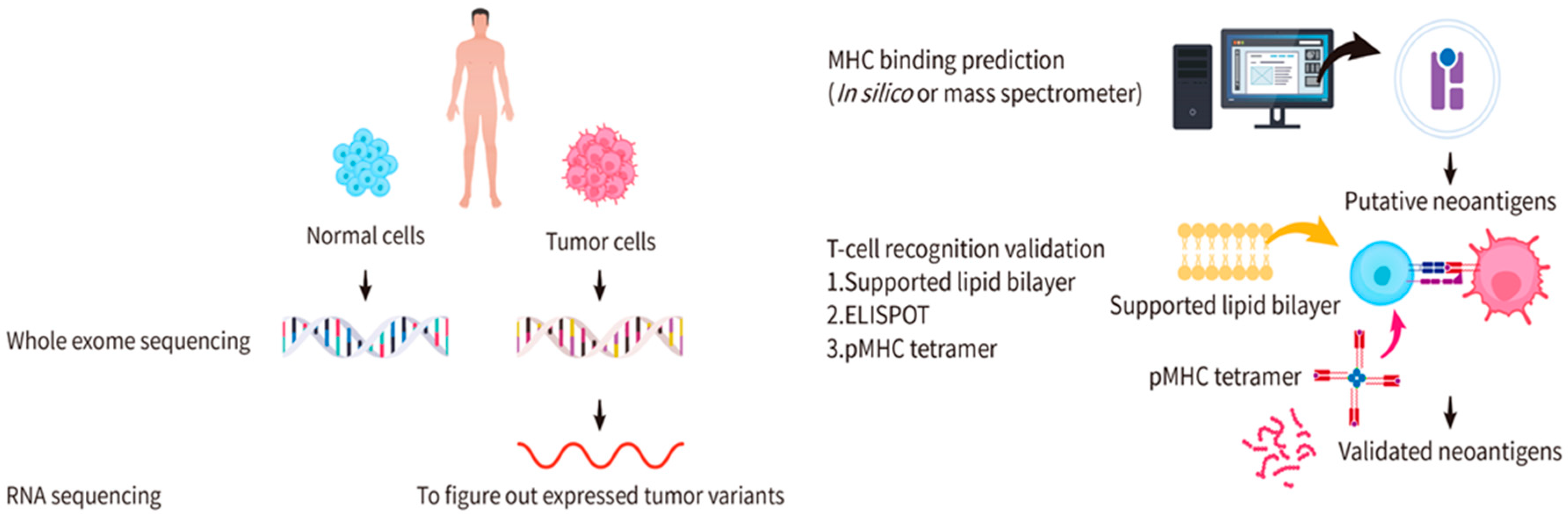
3. Identification and Validation of Candidate Neoantigens
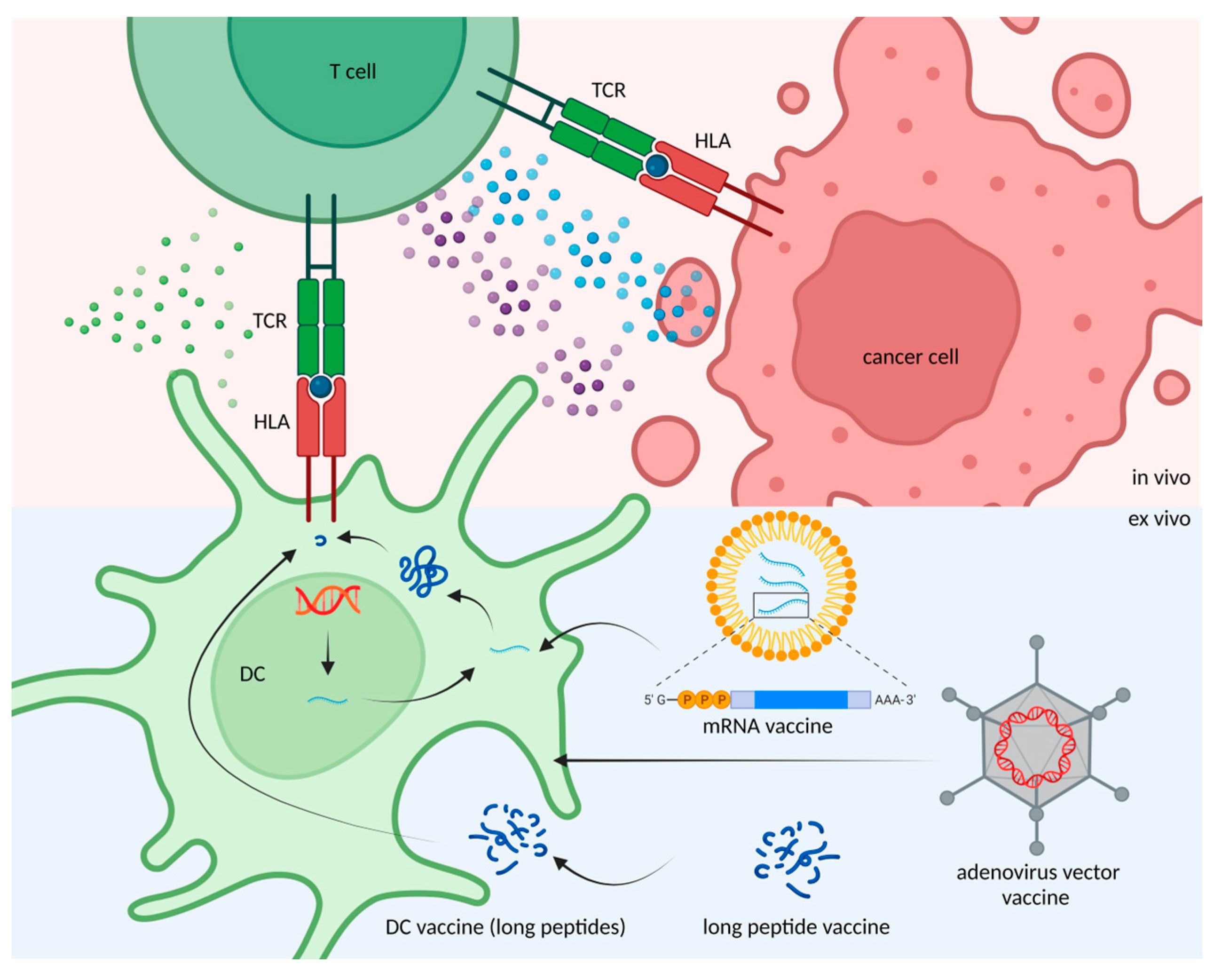
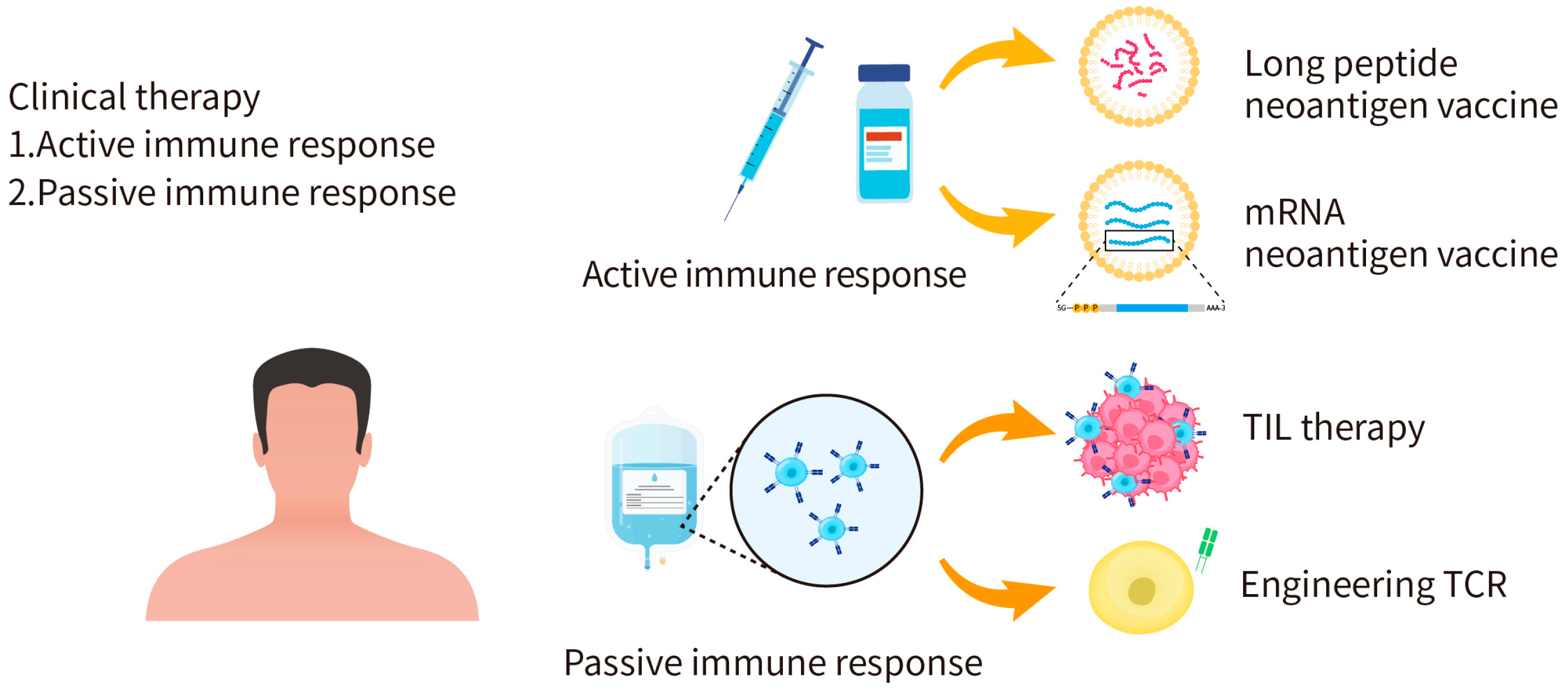
4. Application of Candidate Neoantigen Therapies
5. Challenges
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science: New York, NY, USA, 2016; p. 928. [Google Scholar]
- Kondo, K.; Ohigashi, I.; Takahama, Y. Thymus machinery for T-cell selection. Int. Immunol. 2019, 31, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 2001, 13, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Shoukry, N.H.; Gushima, T.; Bowen, D.G.; Callendret, B.; Campbell, K.J.; Hasselschwert, D.L.; Hughes, A.L.; Walker, C.M. Selection-driven immune escape is not a significant factor in the failure of CD4 T cell responses in persistent hepatitis C virus infection. Hepatology 2010, 51, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, J.; Walker, C.M. Mutational escape of CD8+ T cell epitopes: Implications for prevention and therapy of persistent hepatitis virus infections. Med. Microbiol. Immunol. 2015, 204, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, A.; Saito, T. CD4 CTL, a Cytotoxic Subset of CD4(+) T Cells, Their Differentiation and Function. Front. Immunol. 2017, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N. The cancer antigenome. Embo. J. 2013, 32, 194–203. [Google Scholar] [CrossRef]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Sousa, R.; Keenan, T.E.; Pernas, S.; Exman, P.; Jain, E.; Garrido-Castro, A.C.; Hughes, M.; Bychkovsky, B.; Umeton, R.; Files, J.L.; et al. Tumor Mutational Burden and PTEN Alterations as Molecular Correlates of Response to PD-1/L1 Blockade in Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2565–2572. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.; McGranahan, N.; Rosenthal, R.; et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Momburg, F.; Roelse, J.; Howard, J.C.; Butcher, G.W.; Hämmerling, G.J.; Neefjes, J.J. Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature 1994, 367, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.K.; Holmes, M.A.; Li, P.; Braun, L.; Lee, N.; Geraghty, D.E. HLA-E allelic variants. Correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J. Biol. Chem. 2003, 278, 5082–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minton, K. Immunotherapy: HLA genotype: Good to be different. Nat. Rev. Immunol. 2018, 18, 78–79. [Google Scholar] [CrossRef] [PubMed]
- González, P.A.; Carreño, L.J.; Coombs, D.; Mora, J.E.; Palmieri, E.; Goldstein, B.; Nathenson, S.G.; Kalergis, A.M. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc. Natl. Acad. Sci. USA 2005, 102, 4824–4829. [Google Scholar] [CrossRef] [Green Version]
- Harndahl, M.; Rasmussen, M.; Roder, G.; Dalgaard Pedersen, I.; Sørensen, M.; Nielsen, M.; Buus, S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 2012, 42, 1405–1416. [Google Scholar] [CrossRef]
- Stone, J.D.; Kranz, D.M. Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Front. Immunol. 2013, 4, 244. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef]
- Kristensen, N.P.; Heeke, C.; Tvingsholm, S.A.; Borch, A.; Draghi, A.; Crowther, M.D.; Carri, I.; Munk, K.K.; Holm, J.S.; Bjerregaard, A.M.; et al. Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive cell therapy with tumor-infiltrating lymphocytes in melanoma. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Verdegaal, E.M.; de Miranda, N.F.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.A.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef]
- Holm, J.S.; Funt, S.A.; Borch, A.; Munk, K.K.; Bjerregaard, A.M.; Reading, J.L.; Maher, C.; Regazzi, A.; Wong, P.; Al-Ahmadie, H.; et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 2022, 13, 1935. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Leisegang, M.; Xia, M.; Kiyotani, K.; Li, N.; Zeng, C.; Deng, C.; Jiang, J.; Harada, M.; Agrawal, N.; et al. Generation of neoantigen-specific T cells for adoptive cell transfer for treating head and neck squamous cell carcinoma. Oncoimmunology 2021, 10, 1929726. [Google Scholar] [CrossRef]
- Kvistborg, P.; Clynes, R.; Song, W.; Yuan, J. Immune monitoring technology primer: Whole exome sequencing for neoantigen discovery and precision oncology. J. Immunother. Cancer 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome. Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Gomez-Perosanz, M.; Ras-Carmona, A.; Lafuente, E.M.; Reche, P.A. Identification of CD8(+) T cell epitopes through proteasome cleavage site predictions. BMC Bioinformatics 2020, 21, 484. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, N.; Soban, M.; Chen, F.; Kinkead, H.; Mathew, J.; Yarchoan, M.; Armstrong, T.D.; Haider, S.; Jaffee, E.M. Role of in silico structural modeling in predicting immunogenic neoepitopes for cancer vaccine development. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database-analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The Role of Mass Spectrometry and Proteogenomics in the Advancement of HLA Epitope Prediction. Proteomics 2018, 18, e1700259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, N.; Forsthuber, T.G. ELISPOT Techniques. Methods Mol. Biol. 2016, 1304, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhu, L.; Xu, Q.; Zhang, X.; Li, B.; Lee, L.J. The co-stimulation of anti-CD28 and IL-2 enhances the sensitivity of ELISPOT assays for detection of neoantigen-specific T cells in PBMC. J. Immunol. Methods 2020, 484–485, 112831. [Google Scholar] [CrossRef]
- Lin, J.J.Y.; Low-Nam, S.T.; Alfieri, K.N.; McAffee, D.B.; Fay, N.C.; Groves, J.T. Mapping the stochastic sequence of individual ligand-receptor binding events to cellular activation: T cells act on the rare events. Sci. Signal. 2019, 12, aat8715. [Google Scholar] [CrossRef]
- Natarajan, A.; Krogsgaard, M. The myriad targets of a T cell. Nat. Biotechnol. 2018, 36, 1152–1154. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e324. [Google Scholar] [CrossRef]
- Poran, A.; Scherer, J.; Bushway, M.E.; Besada, R.; Balogh, K.N.; Wanamaker, A.; Williams, R.G.; Prabhakara, J.; Ott, P.A.; Hu-Lieskovan, S.; et al. Combined TCR Repertoire Profiles and Blood Cell Phenotypes Predict Melanoma Patient Response to Personalized Neoantigen Therapy plus Anti-PD-1. Cell Rep. Med. 2020, 1, 100141. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.R.; Al-Batran, S.E.; Tariverdian, M.; Jäger, E.; von Knebel Doeberitz, M. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/IIa Clinical Trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal. Transduct. Target. Ther. 2021, 6, 26. [Google Scholar] [CrossRef]
- Cai, Z.; Su, X.; Qiu, L.; Li, Z.; Li, X.; Dong, X.; Wei, F.; Zhou, Y.; Luo, L.; Chen, G.; et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol. Cancer 2021, 20, 164. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Ellingsen, E.B.; Bounova, G.; Kerzeli, I.; Anzar, I.; Simnica, D.; Aamdal, E.; Guren, T.; Clancy, T.; Mezheyeuski, A.; Inderberg, E.M.; et al. Characterization of the T cell receptor repertoire and melanoma tumor microenvironment upon combined treatment with ipilimumab and hTERT vaccination. J. Transl. Med. 2022, 20, 419. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Govindan, R.; Balogh, K.N.; Spigel, D.R.; Garon, E.B.; Bushway, M.E.; Poran, A.; Sheen, J.H.; Kohler, V.; Esaulova, E.; et al. Personalized neoantigen vaccine NEO-PV-01 with chemotherapy and anti-PD-1 as first-line treatment for non-squamous non-small cell lung cancer. Cancer Cell 2022, 40, 1010–1026.e1011. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.D.; Rappaport, A.R.; Davis, M.J.; Hart, M.G.; Scallan, C.D.; Hong, S.J.; Gitlin, L.; Kraemer, L.D.; Kounlavouth, S.; Yang, A.; et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: Phase 1 trial interim results. Nat. Med. 2022, 28, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef]
- Bunse, L.; Rupp, A.K.; Poschke, I.; Bunse, T.; Lindner, K.; Wick, A.; Blobner, J.; Misch, M.; Tabatabai, G.; Glas, M.; et al. AMPLIFY-NEOVAC: A randomized, 3-arm multicenter phase I trial to assess safety, tolerability and immunogenicity of IDH1-vac combined with an immune checkpoint inhibitor targeting programmed death-ligand 1 in isocitrate dehydrogenase 1 mutant gliomas. Neurol. Res. Pract. 2022, 4, 20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Year | Vaccine | Vaccine Type | Patient Number | Study Phase | Tumor Type | Efficacy | Safety | Trial Identifier |
|---|---|---|---|---|---|---|---|---|
| 2019 [54] | - | Long peptide | 8 | Phase Ib | Glioblastoma | Median progression-free survival: 7.6 months; overall survival: 16.8 months | Grade 1–2 | NCT02287428 |
| 2020 [55] | NEO-PV-01 | Long peptide | 82 advanced melanoma (34), NSCLC (27), bladder cancer (21) | Phase Ib | Advanced melanoma, NSCLC, or bladder cancer | Treated following nivolumab, neoantigen-specific T-cell response was observed in all patients | Mostly Grade 1–2 | NCT02897765 |
| 2020 [56] | NEO-PV-01 | Long peptide | 21 | Phase I | Metastatic melanoma | Prolonged PFS is associated with increased clonal baseline TCR repertoires and longitudinal repertoire stability | - | NCT02897765 |
| 2020 [57] | Micoryx | Frameshift peptide (FSP) neoantigens (mutant AIM2, HT001, and TAF1B genes) | I (6) IIa (16) | Phase I/IIa | DNA mismatch repair (MMR)-deficient colorectal cancer | Vaccine-induced humoral and cellular immune responses were observed in all patients | Grade 1–2 | NCT01461148 |
| 2020 [58] | mRNA-4650 | mRNA vaccine | 4 | Phase I/II | Gastrointestinal cancer | 15.7% of the potential neoantigens induced specific T cell immunity | Grade 1–2 | NCT03480152 |
| 2021 [59] | Neo-DCVac | Dendritic cell vaccines (long peptide) | 12 | Phase I | Advanced lung cancer | Median progression-free survival: 5.5 months; overall survival: 7.9 months | Grade 1–2 | ChiCTR-ONC-16009100, NCT02956551 |
| 2021 [60] | - | Long peptide | 10 | - | Hepatocellular carcinoma | Clinical relapse: 8 patients; relapse-free: 2 patients | Grade 1 | ChiCTR1900020990 |
| 2021 [61] | IDH1-vac | Long peptide | 32 | Phase I | Gliomas with IDH1 mutation | Three-year progression-free rate: 0.63; death-free rate: 0.84. Patients with immune responses showed a 0.82 two-year progression-free rate of 0.82 | Grade 1 | NCT02454634 |
| 2022 [62] | UV1 | Long peptide | 12 | Phase I/IIa | Unresectable metastatic melanoma | Treated in combination with ipilimumab, 91% of evaluable patients showed vaccine-specific immune responses. Clinical responses were observed in four patients (mPFS: 6.7 months, mOS: 66.3 months) | Grade 1–2 | NCT02275416 |
| 2022 [63] | NEO-PV-01 | Long peptide | 38 | Phase Ib | Metastatic non-squamous NSCLC | Treated in combination with anti-PD-1 and chemotherapy, de novo neoantigen-specific CD4+ and CD8+ T-cell responses were observed. Epitope spread to non-vaccinating neoantigens, including responses to KRAS G12C and G12V mutations. | Low grade | NCT03380871 |
| 2022 [64] | - | ChAd68 and samRNA | Fourteen metastatic MSS-CRC (7),GEA (6),NSCLC (1) | Phase 1/2 | Metastatic MSS-CRC, non-small cell lung cancer (NSCLC) and gastroesophageal adenocarcinoma (GEA) | Vaccination combined with nivolumab and ipilimumab induced long-lasting neoantigen-specific CD8 T-cell responses. The median OS rate at 12 months was 8.7 months in MSS-CRC patients. | Mostly Grade 1–2 | NCT03639714 (GRANITE) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, S.-Y.; Chang, C.-M.; Liao, H.-N.; Chou, W.-H.; Guo, C.-L.; Yen, Y.; Nakamura, Y.; Chang, W.-C. Current Trends in Neoantigen-Based Cancer Vaccines. Pharmaceuticals 2023, 16, 392. https://doi.org/10.3390/ph16030392
Ho S-Y, Chang C-M, Liao H-N, Chou W-H, Guo C-L, Yen Y, Nakamura Y, Chang W-C. Current Trends in Neoantigen-Based Cancer Vaccines. Pharmaceuticals. 2023; 16(3):392. https://doi.org/10.3390/ph16030392
Chicago/Turabian StyleHo, Szu-Ying, Che-Mai Chang, Hsin-Ni Liao, Wan-Hsuan Chou, Chin-Lin Guo, Yun Yen, Yusuke Nakamura, and Wei-Chiao Chang. 2023. "Current Trends in Neoantigen-Based Cancer Vaccines" Pharmaceuticals 16, no. 3: 392. https://doi.org/10.3390/ph16030392