
Treatment of Fabry Disease: Established and Emerging Therapies

Abstract
:1. Introduction
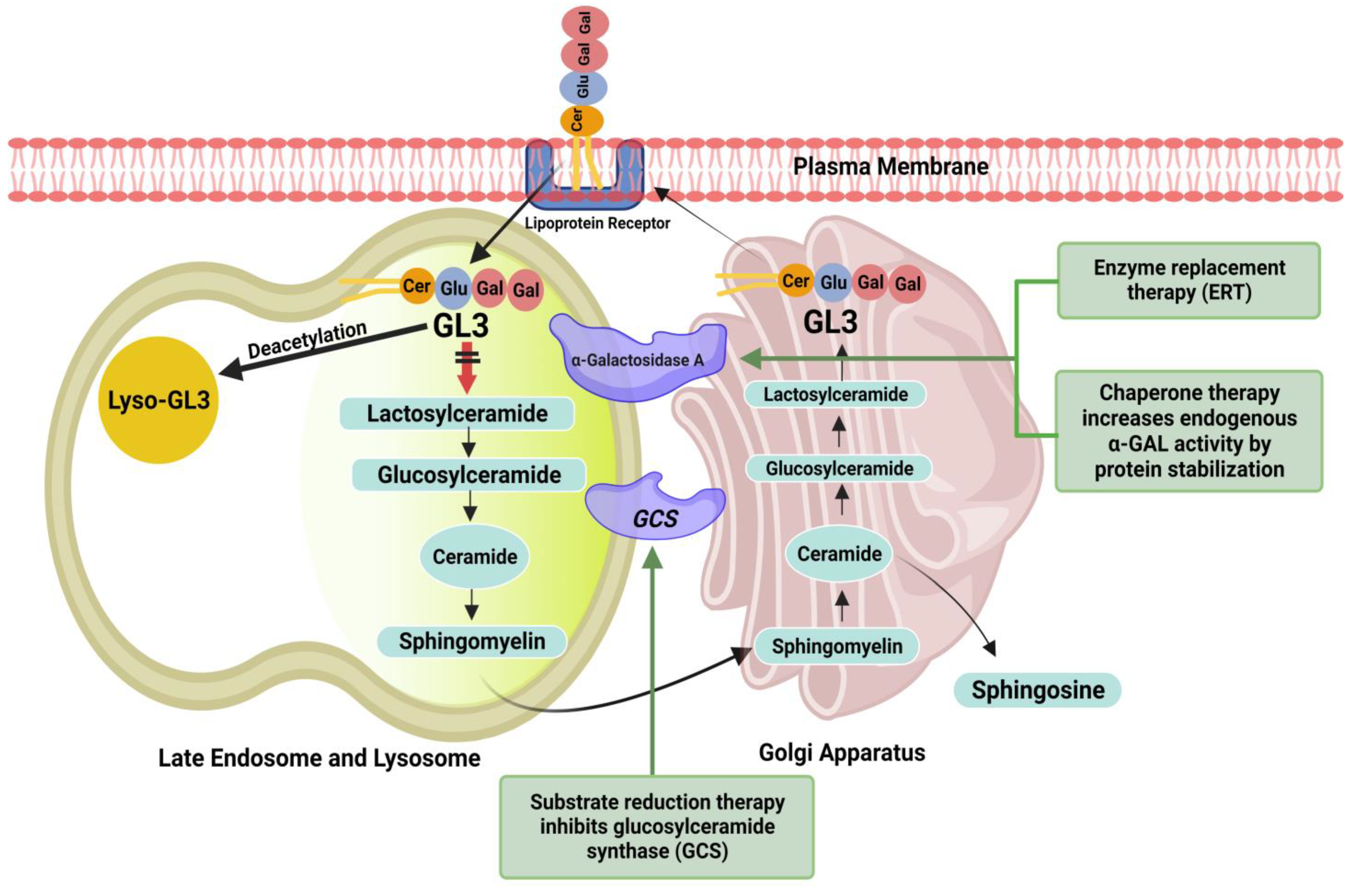
2. Pathophysiology
3. Diagnostic Evaluation
4. Patient Selection and Treatment
4.1. Existing Therapies
4.1.1. Enzyme Replacement Therapy
Agalsidase-β
Agalsidase-α
Anti-Agalsidase Antibody
4.1.2. Chaperone Therapy
Migalastat
4.2. Emerging Therapies
4.2.1. Next Generation ERT
Pegunigalsidase-α
Moss α-GAL
4.2.2. Substrate Reduction
Lucerastat
Venglustat
Genz-682452
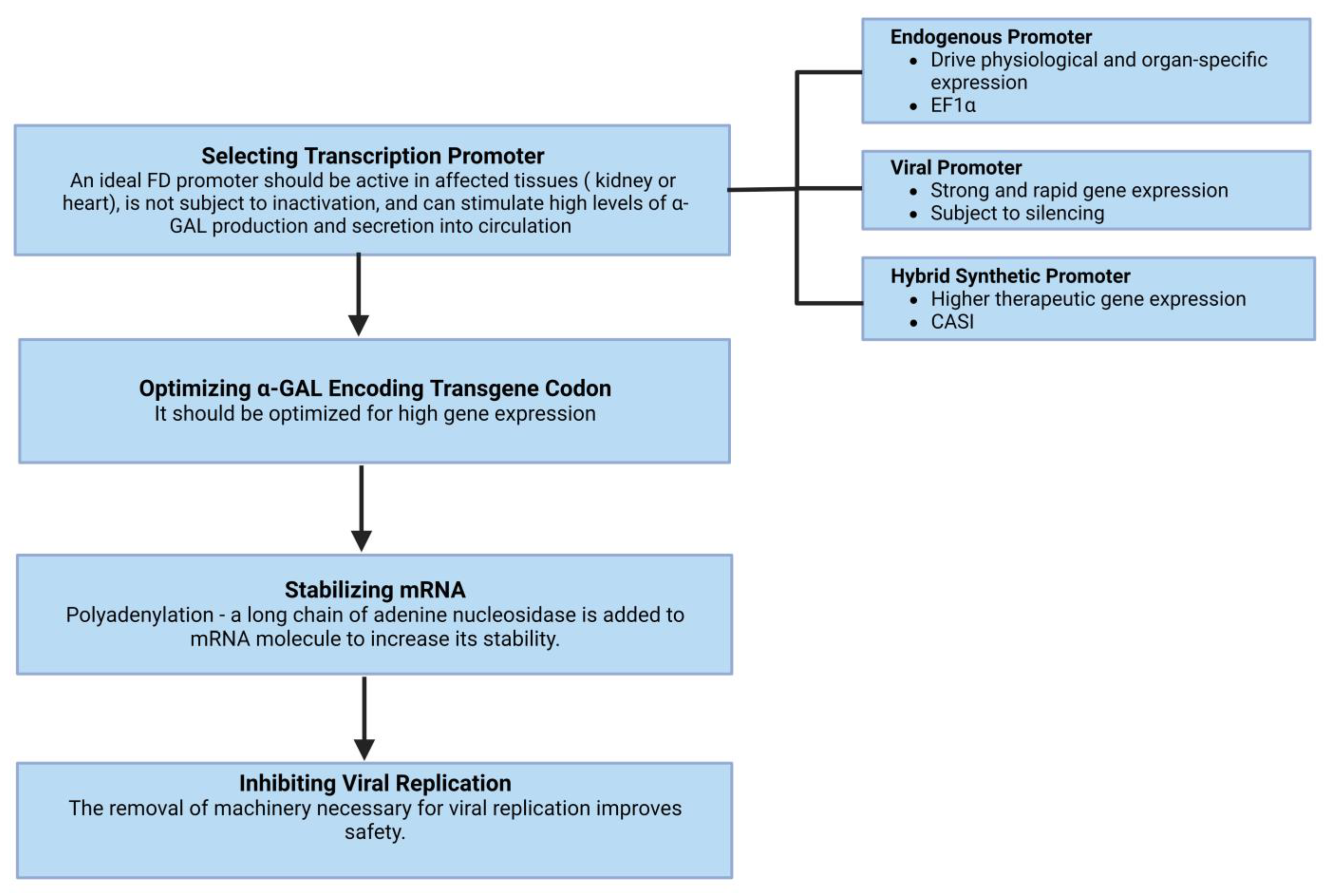
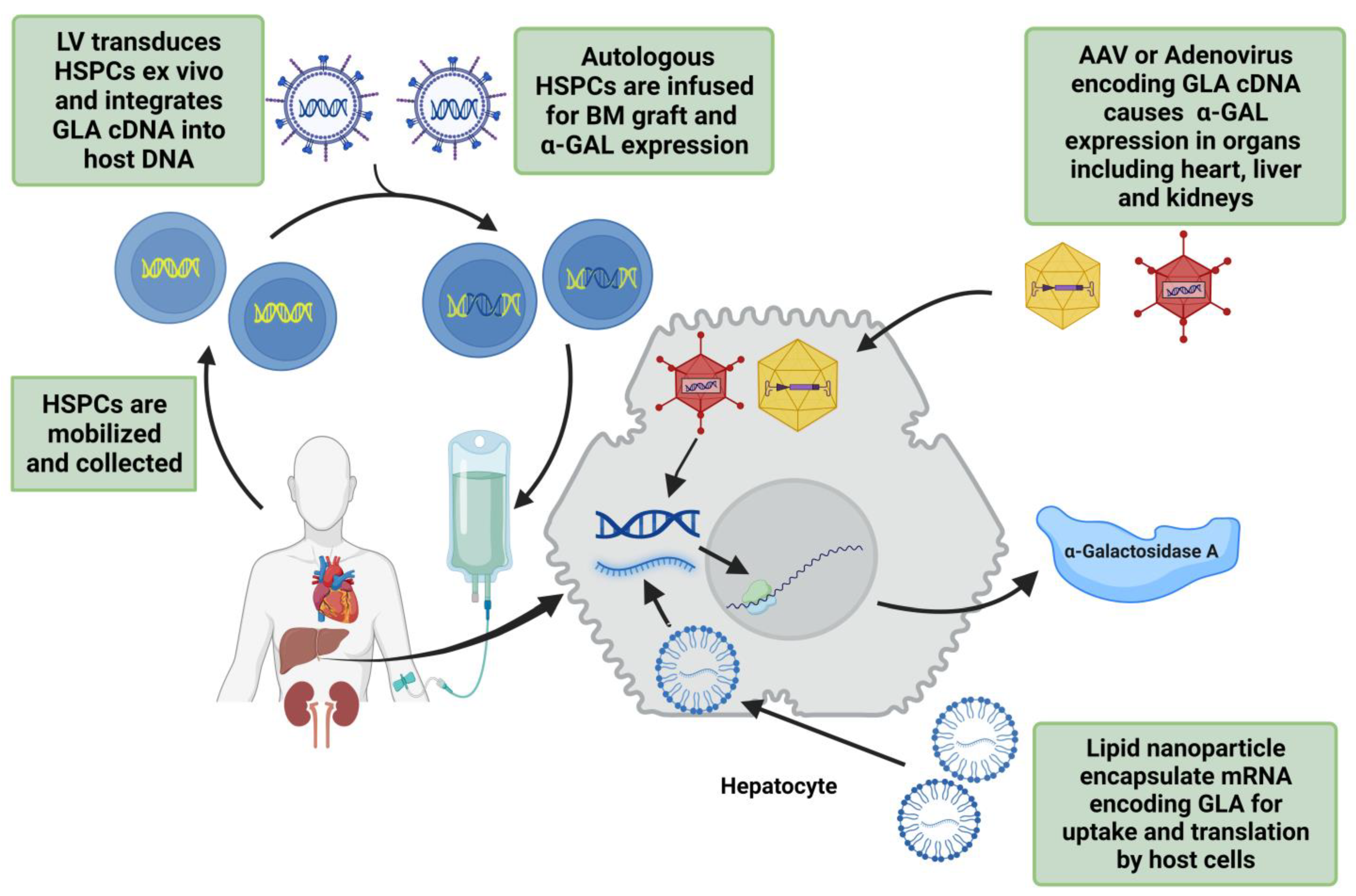
4.2.3. Gene Therapies
Lentivirus
Adenovirus and Adeno-Associated Virus
Plasmid DNA and mRNA
CRISPR (clustered regularly interspaced palindromic repeats)/Cas (CRISPR-associated genes)
4.3. Heart Transplantation
5. Prognosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yousef, Z.; Elliott, P.; Cecchi, F.; Escoubet, B.; Linhart, A.; Monserrat, L.; Namdar, M.; Weidemann, F. Left ventricular hypertrophy in Fabry disease: A practical approach to diagnosis. Eur. Hear. J. 2012, 34, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2011, 58, 2703–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Hughes, D.A. Fabry Disease. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1292/ (accessed on 23 August 2022).
- Eng, C.M.; Fletcher, J.; Wilcox, W.R.; Waldek, S.; Scott, C.R.; Sillence, D.O.; Breunig, F.; Charrow, J.; Germain, D.P.; Nicholls, K.; et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 2007, 30, 184–192. [Google Scholar] [CrossRef]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Anesth. Analg. 2007, 9, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umer, M.; Motwani, M.; Jefferies, J.L.; Nagueh, S.F.; Kalra, D.K. Cardiac Involvement in Fabry Disease and the Role of Multimodality Imaging in Diagnosis and Disease Monitoring. Curr. Probl. Cardiol. 2023, 48. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Fallon, J.T.; Mitchell, R.; Aretz, T.; Gordon, R.E.; O’Callaghan, M.W. Cardiac Microvascular Pathology in Fabry Disease. Circulation 2009, 119, 2561–2567. [Google Scholar] [CrossRef] [Green Version]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction. J. Am. Hear. Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Bachetti, T.; Confortini, R.; Opasich, C.; Febo, O.; Corti, A.; Cassani, G.; Visioli, O. Tumor Necrosis Factor Soluble Receptors in Patients With Various Degrees of Congestive Heart Failure. Circulation 1995, 92, 1479–1486. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.-B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor Necrosis Factor-α and Tumor Necrosis Factor Receptors in the Failing Human Heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef]
- Rauchhaus, M.; Doehner, W.; Francis, D.P.; Davos, C.; Kemp, M.; Liebenthal, C.; Niebauer, J.; Hooper, J.; Volk, H.-D.; Coats, A.S.; et al. Plasma Cytokine Parameters and Mortality in Patients With Chronic Heart Failure. Circulation 2000, 102, 3060–3067. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, C.; Wiethoff, C.M.; Whybra, C.; Baehner, F.A.; Mengel, E.; Beck, M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008, 97, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Herrmann, S.; Hu, K.; Breunig, F.; Strotmann, J.; Beer, M.; Machann, W.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Differences in Fabry Cardiomyopathy Between Female and Male Patients: Consequences for Diagnostic Assessment. JACC Cardiovasc. Imaging 2011, 4, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; De Lorenzo, A.G.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J. Cardiovasc. Magn. Reson. 2017, 19, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, F.; Niemann, M.; Ertl, G.; Störk, S. The Different Faces of Echocardiographic Left Ventricular Hypertrophy: Clues to the Etiology. J. Am. Soc. Echocardiogr. 2010, 23, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Kozor, R.; Callaghan, F.; Tchan, M.; Hamilton-Craig, C.; Figtree, G.A.; Grieve, S.M. A disproportionate contribution of papillary muscles and trabeculations to total left ventricular mass makes choice of cardiovascular magnetic resonance analysis technique critical in Fabry disease. J. Cardiovasc. Magn. Reson. 2015, 17, 22. [Google Scholar] [CrossRef] [Green Version]
- Graziani, F.; Lillo, R.; Panaioli, E.; Spagnoletti, G.; Pieroni, M.; Ferrazzi, P.; Camporeale, A.; Verrecchia, E.; Sicignano, L.L.; Manna, R.; et al. Evidence of evolution towards left midventricular obstruction in severe Anderson–Fabry cardiomyopathy. ESC Hear. Fail. 2020, 8, 725–728. [Google Scholar] [CrossRef]
- Kampmann, C.; Baehner, F.A.; Whybra, C.; Bajbouj, M.; Baron, K.; Knuf, M.; Wiethoff, C.M.; Trübel, H.; Beck, M. The right ventricle in Fabry disease. Acta Paediatr. 2007, 94, 15–18. [Google Scholar] [CrossRef]
- Palecek, T.; Dostalova, G.; Kuchynka, P.; Karetova, D.; Bultas, J.; Elleder, M.; Linhart, A. Right Ventricular Involvement in Fabry Disease. J. Am. Soc. Echocardiogr. 2008, 21, 1265–1268. [Google Scholar] [CrossRef]
- Graziani, F.; Lillo, R.; Panaioli, E.; Pieroni, M.; Camporeale, A.; Verrecchia, E.; Sicignano, L.L.; Manna, R.; Lombardo, A.; Lanza, G.A.; et al. Prognostic significance of right ventricular hypertrophy and systolic function in Anderson–Fabry disease. ESC Hear. Fail. 2020, 7, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Krämer, J.; Niemann, M.; Liu, D.; Hu, K.; Machann, W.; Beer, M.; Wanner, C.; Ertl, G.; Weidemann, F. Two-dimensional speckle tracking as a non-invasive tool for identification of myocardial fibrosis in Fabry disease. Eur. Hear. J. 2013, 34, 1587–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieroni, M.; Chimenti, C.; Ricci, R.; Sale, P.; Russo, M.A.; Frustaci, A. Early Detection of Fabry Cardiomyopathy by Tissue Doppler Imaging. Circulation 2003, 107, 1978–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, L.Z.; Pichette, M.; Nadeau, R.; Bichet, D.G.; Poulin, F. Severe bradyarrhythmia linked to left atrial dysfunction in Fabry disease-A cross-sectional study. Clin. Cardiol. 2018, 41, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Sudhakar, S.; Nanda, N.C.; Buckberg, G.; Pradhan, M.; Roomi, A.U.; Gorissen, W.; Houle, H. Two- and Three-Dimensional Speckle Tracking Echocardiography: Clinical Applications and Future Directions. Echocardiography 2013, 30, 88–105. [Google Scholar] [CrossRef]
- Pieroni, M.; Chimenti, C.; Russo, A.; Russo, M.A.; Maseri, A.; Frustaci, A. Tissue Doppler imaging in Fabry disease. Curr. Opin. Cardiol. 2004, 19, 452–457. [Google Scholar] [CrossRef]
- Liu, D.; Oder, D.; Salinger, T.; Hu, K.; Müntze, J.; Weidemann, F.; Herrmann, S.; Ertl, G.; Wanner, C.; Frantz, S.; et al. Association and diagnostic utility of diastolic dysfunction and myocardial fibrosis in patients with Fabry disease. Open Hear. 2018, 5, e000803. [Google Scholar] [CrossRef]
- Roller, F.C.; Brose, A.; Richter, M.; Schüssler, A.; Harth, S.; Tanislav, C.; Krombach, G.A. Value of Left Ventricular Feature Tracking Strain Analysis for Detection of Early Cardiac Involvement in Fabry Disease (FD). J. Clin. Med. 2021, 10, 3734. [Google Scholar] [CrossRef]
- Vijapurapu, R.; Nordin, S.; Baig, S.; Liu, B.; Rosmini, S.; Augusto, J.; Tchan, M.; Hughes, D.A.; Geberhiwot, T.; Moon, J.; et al. Global longitudinal strain, myocardial storage and hypertrophy in Fabry disease. Hear 2018, 105, 470–476. [Google Scholar] [CrossRef]
- Mathur, S.; Dreisbach, J.G.; Karur, G.R.; Iwanochko, R.M.; Morel, C.F.; Wasim, S.; Nguyen, E.T.; Wintersperger, B.J.; Hanneman, K. Loss of base-to-apex circumferential strain gradient assessed by cardiovascular magnetic resonance in Fabry disease: Relationship to T1 mapping, late gadolinium enhancement and hypertrophy. J. Cardiovasc. Magn. Reson. 2019, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shanks, M.; Thompson, R.; Paterson, D.I.; Putko, B.; Khan, A.; Chan, A.; Becher, H.; Oudit, G.Y. Systolic and Diastolic Function Assessment in Fabry Disease Patients Using Speckle-Tracking Imaging and Comparison with Conventional Echocardiographic Measurements. J. Am. Soc. Echocardiogr. 2013, 26, 1407–1414. [Google Scholar] [CrossRef]
- Deva, D.P.; Hanneman, K.; Li, Q.; Ng, M.Y.; Wasim, S.; Morel, C.; Iwanochko, R.M.; Thavendiranathan, P.; Crean, A.M. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease. J. Cardiovasc. Magn. Reson. 2016, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sado, D.M.; Flett, A.S.; Banypersad, S.M.; White, S.K.; Maestrini, V.; Quarta, G.; Lachmann, R.; Murphy, E.; Mehta, A.; Hughes, D.; et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Hear 2012, 98, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Vijapurapu, R.; Augusto, J.; Knott, K.D.; Captur, G.; Treibel, T.; Ramaswami, U.; Tchan, M.; Geberhiwot, T.; et al. Myocardial Storage, Inflammation, and Cardiac Phenotype in Fabry Disease After One Year of Enzyme Replacement Therapy. Circ. Cardiovasc. Imaging 2019, 12, e009430. [Google Scholar] [CrossRef]
- Nordin, S.; Kozor, R.; Bulluck, H.; Castelletti, S.; Rosmini, S.; Abdel-Gadir, A.; Baig, S.; Mehta, A.; Hughes, D.; Moon, J. Cardiac Fabry Disease With Late Gadolinium Enhancement Is a Chronic Inflammatory Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 1707–1708. [Google Scholar] [CrossRef]
- Hughes, D.A.; Elliott, P.; Shah, J.; Zuckerman, J.; Coghlan, G.; Brookes, J.; Mehta, A.B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson Fabry disease: A randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Hear 2008, 94, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Turschner, O.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. Improvement of Cardiac Function During Enzyme Replacement Therapy in Patients With Fabry Disease. Circulation 2003, 108, 1299–1301. [Google Scholar] [CrossRef] [Green Version]
- Wuest, W.; Machann, W.; Breunig, F.; Weidemann, F.; Koestler, H.; Hahn, D.; Wanner, C.; Beer, M. Right Ventricular Involvement in Patients with Fabry’s Disease and the Effect of Enzyme Replacement Therapy. Rofo 2011, 183, 1037–1042. [Google Scholar] [CrossRef]
- Spinelli, L.; Pisani, A.; Sabbatini, M.; Petretta, M.; Andreucci, M.; Procaccini, D.; Surdo, N.L.; Federico, S.; Cianciaruso, B. Enzyme replacement therapy with agalsidase β improves cardiac involvement in Fabry’s disease. Clin. Genet. 2004, 66, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Stoörk, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-Term Effects of Enzyme Replacement Therapy on Fabry Cardiomyopathy. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.; Bijnens, B.; Störk, S.; Ritter, C.O.; Liu, D.; Ertl, G.; Wanner, C.; Weidemann, F. Left Ventricular Geometry and Blood Pressure as Predictors of Adverse Progression of Fabry Cardiomyopathy. PLoS ONE 2015, 10, e0140627. [Google Scholar] [CrossRef] [PubMed]
- Imbriaco, M.; Pisani, A.; Spinelli, L.; Cuocolo, A.; Messalli, G.; Capuano, E.; Marmo, M.; Liuzzi, R.; Visciano, B.; Cianciaruso, B.; et al. Effects of enzyme-replacement therapy in patients with Anderson-Fabry disease: A prospective long-term cardiac magnetic resonance imaging study. Hear 2009, 95, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Messalli, G.; Imbriaco, M.; Avitabile, G.; Russo, R.; Iodice, D.; Spinelli, L.; Dellegrottaglie, S.; Cademartiri, F.; Salvatore, M.; Pisani, A. Role of cardiac MRI in evaluating patients with Anderson-Fabry disease: Assessing cardiac effects of long-term enzyme replacement therapy. La Radiol. Med. 2011, 117, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Hopkin, R.J.; Jefferies, J.L.; Laney, D.A.; Lawson, V.H.; Mauer, M.; Taylor, M.R.; Wilcox, W.R.; Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol. Genet. Metab. 2016, 117, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Arngrímsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Iii, H.A.A.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Tallman, J.F.; Johnson, W.G.; Gal, A.E.; Leahy, W.R.; Quirk, J.M.; Dekaban, A.S. Replacement Therapy for Inherited Enzyme Deficiency. N. Engl. J. Med. 1973, 289, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, S.; Neubauer, H.; Breunig, F.; Weidemann, F.; Wanner, C.; Sandstede, J.; Machann, W.; Hahn, D.; Köstler, H.; Beer, M. MR-based analysis of regional cardiac function in relation to cellular integrity in Fabry disease. Int. J. Cardiol. 2011, 160, 53–58. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.-G.; Warnock, D.G.; et al. Agalsidase-Beta Therapy for Advanced Fabry Disease. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Tylki-Szymanska, A.; Guffon, N.; Lien, Y.H.; Tsimaratos, M.; Vellodi, A.; Germain, D.P. Safety and Efficacy of Enzyme Replacement Therapy with Agalsidase Beta: An International, Open-label Study in Pediatric Patients with Fabry Disease. J. Pediatr. 2008, 152, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Dormond, O.; Monzambani, V.; Huynh-Do, U.; Barbey, F. Agalsidase-β should be proposed as first line therapy in classic male Fabry patients with undetectable α-galactosidase A activity. Mol. Genet. Metab. 2022, 137, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Pastores, G.M.; Lien, Y.-H.H.; Castaneda, V.; Chang, P.; Martin, R.; Wijatyk, A. Agalsidase alfa in pediatric patients with Fabry disease: A 6.5-year open-label follow-up study. Orphanet J. Rare Dis. 2014, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, K.; Yamamoto, H. Clinical course of patients with Fabry disease who were switched from agalsidase-β to agalsidase-α. Anesthesia Analg. 2014, 16, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Mauhin, W.; Lidove, O.; Amelin, D.; Lamari, F.; Caillaud, C.; Mingozzi, F.; Dzangué-Tchoupou, G.; Arouche-Delaperche, L.; Douillard, C.; Dussol, B.; et al. Deep characterization of the anti-drug antibodies developed in Fabry disease patients, a prospective analysis from the French multicenter cohort FFABRY. Orphanet J. Rare Dis. 2018, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Stypmann, J.; Duning, T.; Schmitz, B.; Brand, S.-M.; Brand, E. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J. Am. Soc. Nephrol. 2016, 27, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Rombach, S.M.; Aerts, J.; Poorthuis, B.J.H.M.; Groener, J.E.M.; Donker-Koopman, W.; Hendriks, E.; Mirzaian, M.; Kuiper, S.; Wijburg, F.A.; Hollak, C.E.M.; et al. Long-Term Effect of Antibodies against Infused Alpha-Galactosidase A in Fabry Disease on Plasma and Urinary (lyso)Gb3 Reduction and Treatment Outcome. PLoS ONE 2012, 7, e47805. [Google Scholar] [CrossRef]
- GALAFOLD® GLA Mutation Search. Available online: https://galafoldamenabilitytable.com/hcp (accessed on 23 August 2022).
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Anesth. Analg. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Eveslage, M.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; et al. Treatment of Fabry Disease management with migalastat—Outcome from a prospective 24 months observational multicenter study (FAMOUS). Eur. Hear. J. Cardiovasc. Pharmacother. 2021, 8, 272–281. [Google Scholar] [CrossRef]
- Hughes, D.A.; Bichet, D.G.; Giugliani, R.; Hopkin, R.J.; Krusinska, E.; Nicholls, K.; Olivotto, I.; Feldt-Rasmussen, U.; Sakai, N.; Skuban, N.; et al. Long-term multisystemic efficacy of migalastat on Fabry-associated clinical events, including renal, cardiac and cerebrovascular outcomes. J. Med. Genet. 2022. [Google Scholar] [CrossRef]
- Schiffmann, R.; Goker-Alpan, O.; Holida, M.; Giraldo, P.; Barisoni, L.; Colvin, R.B.; Jennette, C.J.; Maegawa, G.; Boyadjiev, S.A.; Gonzalez, D.; et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Hennermann, J.B.; Arash-Kaps, L.; Fekete, G.; Schaaf, A.; Busch, A.; Frischmuth, T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J. Inherit. Metab. Dis. 2019, 42, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guérard, N.; Oder, D.; Nordbeck, P.; Zwingelstein, C.; Morand, O.; Welford, R.W.; Dingemanse, J.; Wanner, C. Lucerastat, an Iminosugar for Substrate Reduction Therapy: Tolerability, Pharmacodynamics, and Pharmacokinetics in Patients With Fabry Disease on Enzyme Replacement. Clin. Pharmacol. Ther. 2017, 103, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 10, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Goker-Alpan, O.; Geberhiwot, T.; Hopkin, R.J.; Lukina, E.; Tylki-Szymanska, A.; Zaher, A.; Sensinger, C.; Gaemers, S.J.; Modur, V.; et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: Assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol. Genet. Metab. 2023, 138, 106963. [Google Scholar] [CrossRef]
- Ashe, K.M.; Budman, E.; Bangari, D.S.; Siegel, C.S.; Nietupski, J.B.; Wang, B.; Desnick, R.J.; Scheule, R.K.; Leonard, J.P.; Cheng, S.H.; et al. Efficacy of Enzyme and Substrate Reduction Therapy with a Novel Antagonist of Glucosylceramide Synthase for Fabry Disease. Mol. Med. 2015, 21, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Qin, G.; Brady, R.O.; Medin, J.A. Circulating alpha-Galactosidase A Derived from Transduced Bone Marrow Cells: Relevance for Corrective Gene Transfer for Fabry Disease. Hum. Gene Ther. 1999, 10, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Medin, J.A.; Tudor, M.; Simovitch, R.; Quirk, J.M.; Jacobson, S.; Murray, G.J.; Brady, R.O. Correction in trans for Fabry disease: Expression, secretion and uptake of alpha-galactosidase A in patient-derived cells driven by a high-titer recombinant retroviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 7917–7922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, T.; Murray, G.J.; Qin, G.; Quirk, J.M.; Ohshima, T.; Qasba, P.; Clark, K.; Kulkarni, A.B.; Brady, R.O.; Medin, J.A. Long-term enzyme correction and lipid reduction in multiple organs of primary and secondary transplanted Fabry mice receiving transduced bone marrow cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7515–7520. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, M.; Higuchi, K.; Ramsubir, S.; Nonaka, T.; Rasaiah, V.I.; Siatskas, C.; Liang, S.-B.; Murray, G.J.; Brady, R.O.; Medin, J.A. Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 2006, 14, 256–265. [Google Scholar] [CrossRef]
- Pacienza, N.; Yoshimitsu, M.; Mizue, N.; Au, B.C.; Wang, J.C.; Fan, X.; Takenaka, T.; Medin, J.A. Lentivector Transduction Improves Outcomes Over Transplantation of Human HSCs Alone in NOD/SCID/Fabry Mice. Mol. Ther. 2012, 20, 1454–1461. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef]
- Ziegler, R.J.; Yew, N.S.; Li, C.; Cherry, M.; Berthelette, P.; Romanczuk, H.; Ioannou, Y.A.; Zeidner, K.M.; Desnick, R.J.; Cheng, S.H. Correction of Enzymatic and Lysosomal Storage Defects in Fabry Mice by Adenovirus-Mediated Gene Transfer. Hum. Gene Ther. 1999, 10, 1667–1682. [Google Scholar] [CrossRef]
- Ziegler, R.J.; Li, C.; Cherry, M.; Zhu, Y.; Hempel, D.; Van Rooijen, N.; Ioannou, Y.A.; Desnick, R.J.; Goldberg, M.A.; Yew, N.S.; et al. Correction of the Nonlinear Dose Response Improves the Viability of Adenoviral Vectors for Gene Therapy of Fabry Disease. Hum. Gene Ther. 2002, 13, 935–945. [Google Scholar] [CrossRef]
- Reid, T.; Warren, R.S.; Kirn, D.H. Intravascular adenoviral agents in cancer patients: Lessons from clinical trials. Cancer Gene Ther. 2002, 9, 979–986. [Google Scholar] [CrossRef]
- George, L.A.; Ragni, M.V.; Rasko, J.E.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K.; et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotterman, M.; Whittlesey, K.; Brooks, G.; Croze, R.; Schmitt, C.; Szymanski, P.; Nye, J.; Quezada, M.; Beliakoff, G.; Johnson, L.; et al. P1516Novel cardiotropic AAV variant C102 vectors show superior gene delivery & reduced immunogenicity in non-human primates, transduction of human cardiomyocytes, & correction of Fabry disease phenotype. Eur. Hear. J. 2019, 40. [Google Scholar] [CrossRef]
- Jung, S.-C.; Han, I.P.; Limaye, A.; Xu, R.; Gelderman, M.P.; Zerfas, P.; Tirumalai, K.; Murray, G.J.; During, M.J.; Brady, R.O.; et al. Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2676–2681. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Hirai, Y.; Migita, M.; Seino, Y.; Fukuda, Y.; Sakuraba, H.; Kase, R.; Kobayashi, T.; Hashimoto, Y.; Shimada, T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 13777–13782. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Murray, G.J.; Limaye, A.; Quirk, J.M.; Gelderman, M.P.; Brady, R.O.; Qasba, P. Long-term correction of globotriaosylceramide storage in Fabry mice by recombinant adeno-associated virus-mediated gene transfer. Proc. Natl. Acad. Sci. USA 2003, 100, 3450–3454. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, R.J.; Lonning, S.M.; Armentano, D.; Li, C.; Souza, D.W.; Cherry, M.; Ford, C.; Barbon, C.M.; Desnick, R.J.; Gao, G.; et al. AAV2 Vector Harboring a Liver-Restricted Promoter Facilitates Sustained Expression of Therapeutic Levels of α-Galactosidase A and the Induction of Immune Tolerance in Fabry Mice. Mol. Ther. 2004, 9, 231–240. [Google Scholar] [CrossRef]
- van Lieshout, L.P.; Domm, J.M.; Rindler, T.N.; Frost, K.L.; Sorensen, D.L.; Medina, S.J.; Booth, S.A.; Bridges, J.; Wootton, S.K. A Novel Triple-Mutant AAV6 Capsid Induces Rapid and Potent Transgene Expression in the Muscle and Respiratory Tract of Mice. Mol. Ther. Methods Clin. Dev. 2018, 9, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming Preexisting Humoral Immunity to AAV Using Capsid Decoys. Sci. Transl. Med. 2013, 5, 194ra92. [Google Scholar] [CrossRef] [Green Version]
- Halbert, C.L.; Standaert, T.A.; Wilson, C.B.; Miller, A.D. Successful Readministration of Adeno-Associated Virus Vectors to the Mouse Lung Requires Transient Immunosuppression during the Initial Exposure. J. Virol. 1998, 72, 9795–9805. [Google Scholar] [CrossRef] [Green Version]
- Elmore, Z.C.; Oh, D.K.; Simon, K.E.; Fanous, M.M.; Asokan, A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. J. Clin. Investig. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Metzger, M.J.; Lam, S.-L.; Miller, A.D. Capsid-expressing DNA in AAV vectors and its elimination by use of an oversize capsid gene for vector production. Gene Ther. 2010, 18, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.-K.; Lu, Y.-H.; Chen, Y.-R.; Hsieh, Y.-P.; Lin, W.-J.; Hsu, T.-R.; Niu, D.-M. AB043. Correction of the GLA IVS4+919 G>A mutation with CRISPR/Cas9 deletion strategy in fibroblasts of Fabry disease. Ann. Transl. Med. 2017, 5, AB043. [Google Scholar] [CrossRef] [Green Version]
- Di Nora, C.; Livi, U. Heart transplantation in cardiac storage diseases: Data on Fabry disease and cardiac amyloidosis. Curr. Opin. Organ. Transplant. 2020, 25, 211–217. [Google Scholar] [CrossRef]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 2001, 38, 750–760. [Google Scholar] [CrossRef] [Green Version]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS - Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsborne, C.; Bradley, J.; Bonnett, L.J.; Pleva, L.A.; Naish, J.H.; Clark, D.G.; Abidin, N.; Woolfson, P.; Nucifora, G.; Schmitt, M.; et al. Validated Model for Prediction of Adverse Cardiac Outcome in Patients With Fabry Disease. J. Am. Coll. Cardiol. 2022, 80, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Hanneman, K.; Karur, G.R.; Wasim, S.; Wald, R.M.; Iwanochko, R.M.; Morel, C.F. Left Ventricular Hypertrophy and Late Gadolinium Enhancement at Cardiac MRI Are Associated with Adverse Cardiac Events in Fabry Disease. Radiology 2020, 294, 42–49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Structural Evaluation | Functional Evaluation | Tissue Characterization | ||||
|---|---|---|---|---|---|---|
| Ventricles and Atria | Diastolic Dysfunction | Systolic Dysfunction | Tissue Doppler Imaging Strain Analysis | LGE Pattern | T1 Mapping Extracellular Volume | T2 Mapping |
| The majority of the patients have concentric LV wall thickening [17]. Papillary muscle hypertrophy can contribute to the LV mass and cause mid-ventricular obstruction [18,19]. RV involvement is present in 31–71% of the patients, causing RV hypertrophy with normal chamber size [20,21]. RVH and RV systolic function show significant association with clinical events, but RV involvement does not influence prognosis [22]. Atrial dilatation is more common in patients with LVH and fibrosis [23,24], and left atrial dysfunction is associated with Bradyarrhythmia [25]. | LV diastolic dysfunction may occur before the development of LVH [26]. RV diastolic dysfunction can develop with advancing left ventricular cardiomyopathy. | LV systolic dysfunction does not usually occur until the late stages of cardiac involvement. Despite RV hypertrophy and fibrosis, RV systolic function is generally preserved. | TDI is an accurate and sensitive tool for diagnosing myocardial dysfunction, even before the development of LVH [24,27]. Septal E/e’ can predict replacement fibrosis in the absence of LVH and diastolic dysfunction [28]. GLS and GRS are significantly reduced, and GLS impairment correlates with GL3/lyso-GL3 elevation, thus having a potential role in detecting early cardiac involvement [29,30]. Loss of base-to-apex CS gradient is also an early marker of cardiac involvement [31]. Longitudinal systolic strain, systolic strain rate, and longitudinal early diastolic strain rate are superior TTE measures of LV function and are reduced independent of LVH [32]. Reduced longitudinal strain in the basal inferolateral wall can help differentiate from other hypertrophic cardiomyopathies [23]. | Basal to mid inferolateral mid-myocardium [33]. | Initially, native T1 values are reduced, but later there is pseudonormalization in the areas of LGE. It can reliably distinguish FD from other causes of LVH (27). ECV is normal [34] but may increase in the area of LGE as a biomarker for fibrosis. | T2 values are elevated in the area of LGE, indicating chronic inflammation [35,36]. |
| Monitoring of treatment efficacy | ||||||
| Ventricles and Atria | Diastolic dysfunction | Systolic dysfunction | Tissue doppler imaging Strain analysis | LGE pattern | T1 mapping Extracellular volume | T2 mapping |
| LVM regression occurs in patients with baseline LVH. Patients with little or no LGE at baseline also showed a decrease in LVM [35,37,38]. LVM reduction varies among various studies (10–27%), likely depending on the timing of therapy, the intensity of therapy, the stage of cardiomyopathy, and other confounding factors, such as age, sex, hypertension, etc. RV mass may decrease with ERT [39]. | No change [38] or very minor improvement in diastolic dysfunction is noted with ERT [40]. | Radial and longitudinal function of LV improves after 12 months of ERT, especially in the last 6 months of treatment [38]. Patients without fibrosis showed improvement in systolic function over 3 years on ERT, although no improvement was noted in patients with mild or severe fibrosis [41]. | Significant increase of peak systolic strain rate (radial function, baseline, 2.8 ± 0.2 s−1; 12 months, 3.7 ± 0.3 s−1; p < 0.05) and end-systolic strain (baseline, 34 ± 3%; 12 months, 45 ± 4%; p < 0.05) is noted in the posterior wall after 12 months of ERT [38]. Patients without fibrosis showed improvement in systolic radial strain rate (2.3 ± 0.4 to 2.9 ± 0.6 s−1; p = 0.045) over a long-term follow-up of 3 years on ERT. No improvement in patients with mild or severe fibrosis [41]. LV inferolateral regional strain can predict the progression of LGE [42]. | No improvement is noted with treatment. | Reduction in T1 lowering with ERT [35]. | Decrease in T2 due to a reduction in myocardial lipid burden [43,44]. |
| Drug Name | Mechanism of Action | Route of Administration | Physiological Effect |
|---|---|---|---|
| Agalsidase-β | Recombinant α-GAL | Intravenous infusion | Decreases accumulation of GL3 |
| Agalsidase-α | Recombinant α-GAL | Intravenous infusion | Decreases accumulation of GL3 |
| Migalastat | Binds reversibly to the active site of the amenable mutant of α-GAL | Oral | Promotes trafficking of α-GAL to lysosome, thus increasing enzyme activity |
| Investigational drugs | |||
| Pegunigalsidase-α | Plant derived α-GAL | Intravenous infusion | Decreases accumulation of GL3 |
| Moss α-GAL | Moss derived α-GAL | Intravenous infusion | Decreases accumulation of GL3 |
| Lucerastat | Glucosylceramide synthase inhibitor | Oral | Reduces accumulation of glycosphingolipids, including GL1 and GL3 |
| Venglustat | Glucosylceramide synthase inhibitor | Oral | Reduces accumulation of glycosphingolipids, including GL1 and GL3 |
| Initiation of ERT | Discontinuation of ERT | Not a Candidate for ERT |
|---|---|---|
| Classical FD males: 16 years or older, even if they have no symptoms or clinical signs of organ involvement (Class IIB recommendation) Classical FD males and females: as soon as there are early signs of FD organ involvement (kidney, heart, and/or CNS signs) and not fully explained by other pathology (Class I recommendation). Non-classical FD males: as soon as there are early signs of FD organ involvement (kidney, heart, and/or CNS signs and not fully explained by other pathology (Class I recommendation). Non-classical FD females: early clinical signs consistent with FD (Class IIB recommendation) Cardiac-specific criteria: myocardial hypertrophy (MWT > 12 mm) without (or only minimal signs of) fibrosis (Class I) or signs of cardiac rhythm disturbances, including sinus bradycardia, AF, and repolarization disorders (Class I) | Noncompliance with >50 percent of treatments or patient request (Class 1). End-stage renal disease, without an option for renal transplantation, in combination with advanced heart failure (NYHA class IV)–Class IIA End-stage FD or other comorbidities with a life expectancy of <1 year-Class IIB The severe cognitive decline of any cause-Class IIB Lack of response for 1 year when the sole indication for ERT is neuropathic pain while receiving maximum supportive care. An exception is classical FD males who are at high risk of developing clinical signs of organ involvement within a short time period (Class II B). | Advanced cardiac disease with extensive fibrosis-Class I End-stage renal disease, without an option for renal transplantation, in combination with advanced heart failure (NYHA class IV)-Class IIA End-stage FD or other comorbidities with a life expectancy of <1 year-Class IIB The severe cognitive decline of any cause-Class IIB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umer, M.; Kalra, D.K. Treatment of Fabry Disease: Established and Emerging Therapies. Pharmaceuticals 2023, 16, 320. https://doi.org/10.3390/ph16020320
Umer M, Kalra DK. Treatment of Fabry Disease: Established and Emerging Therapies. Pharmaceuticals. 2023; 16(2):320. https://doi.org/10.3390/ph16020320
Chicago/Turabian StyleUmer, Muhammad, and Dinesh K. Kalra. 2023. "Treatment of Fabry Disease: Established and Emerging Therapies" Pharmaceuticals 16, no. 2: 320. https://doi.org/10.3390/ph16020320