
PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer

Abstract
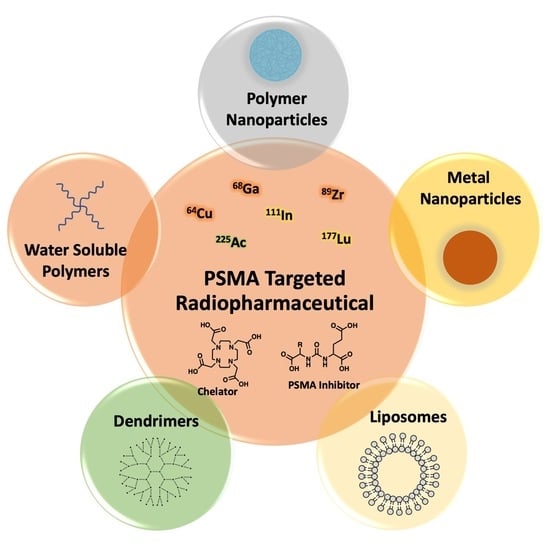
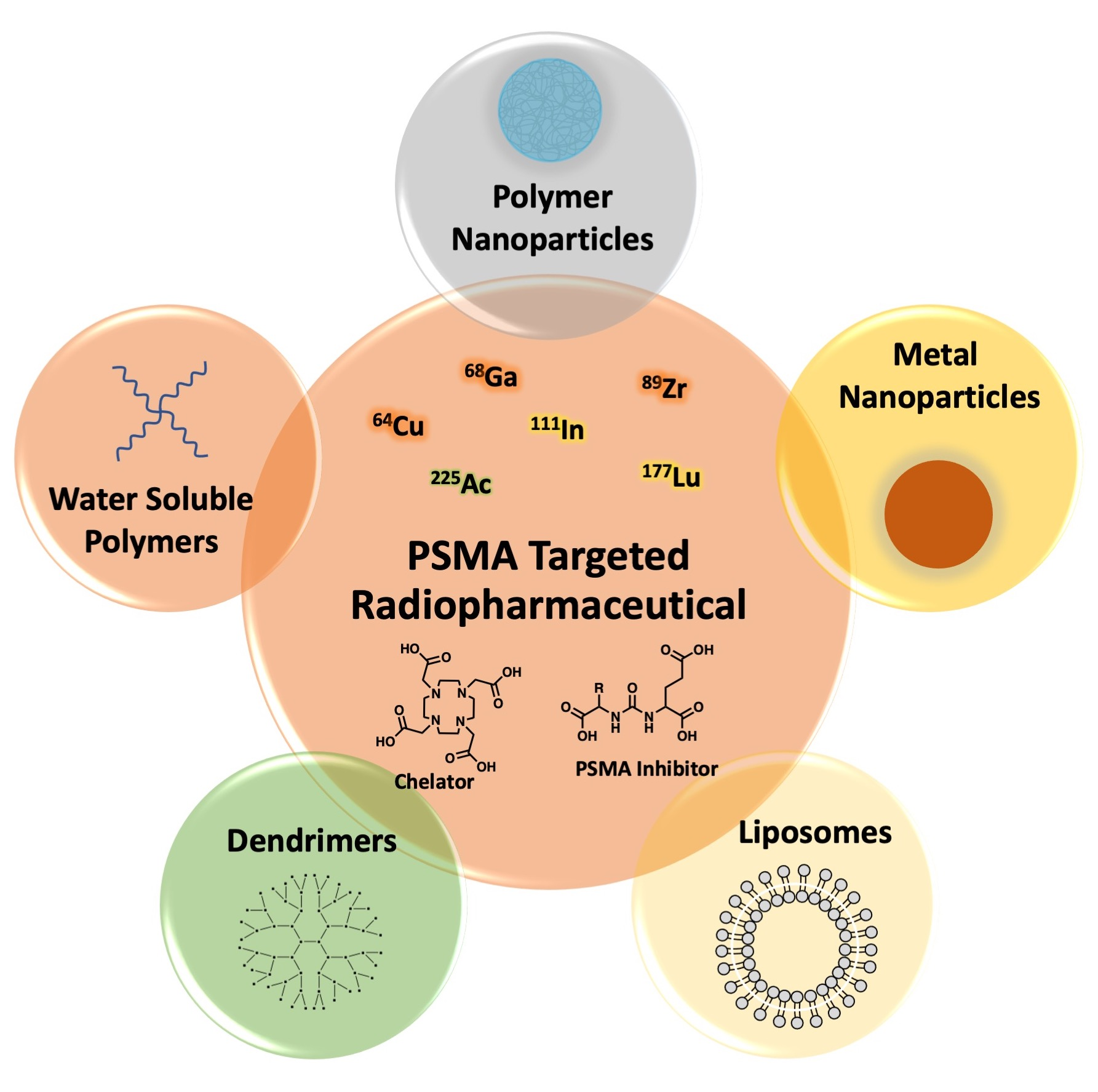
:
1. Introduction
2. Radiometals and Chelators for Imaging and Therapy
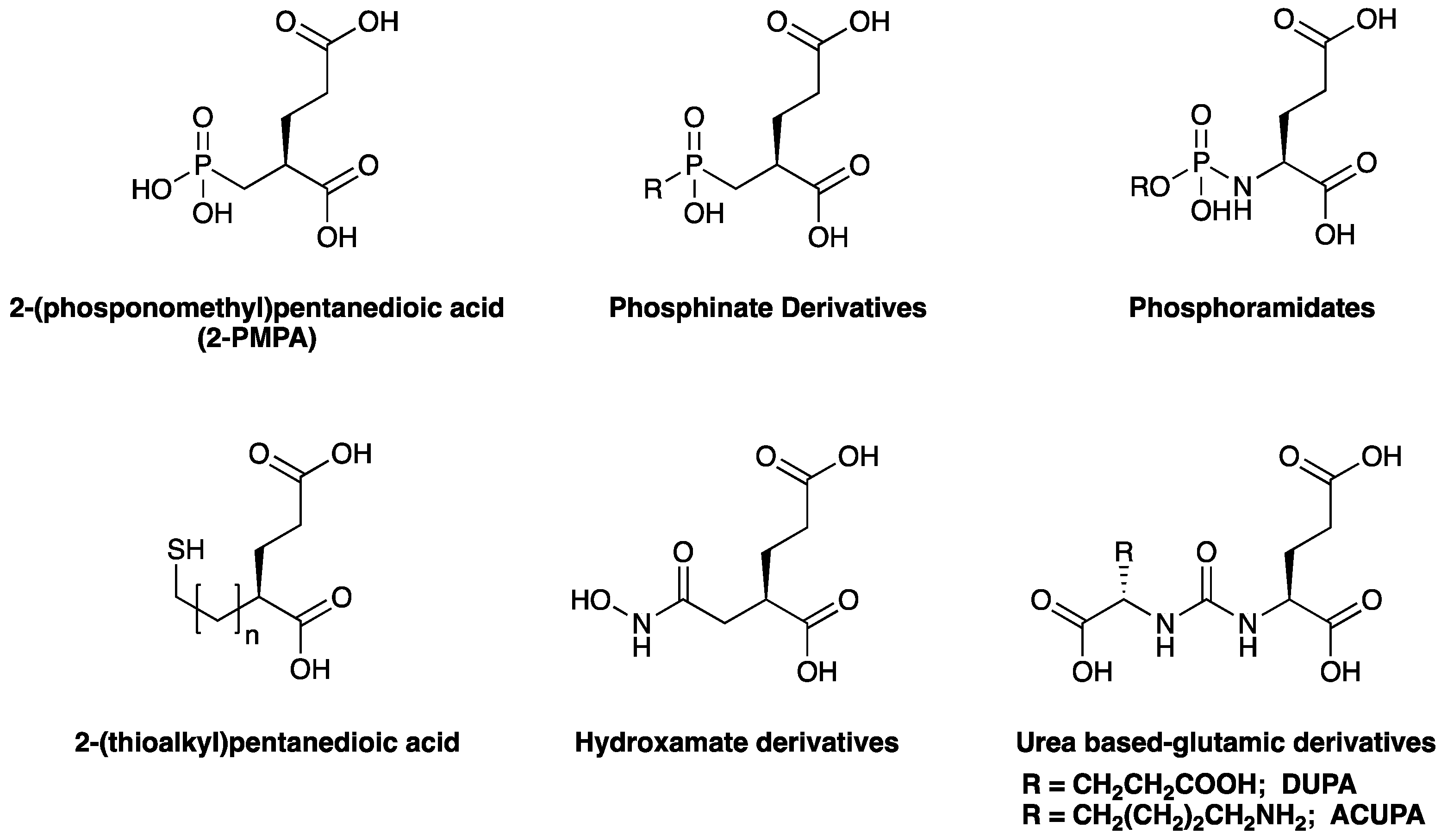
3. PSMA and Its Targeting Ligands
4. EPR Effect and Need of Targeted Nanomedicine
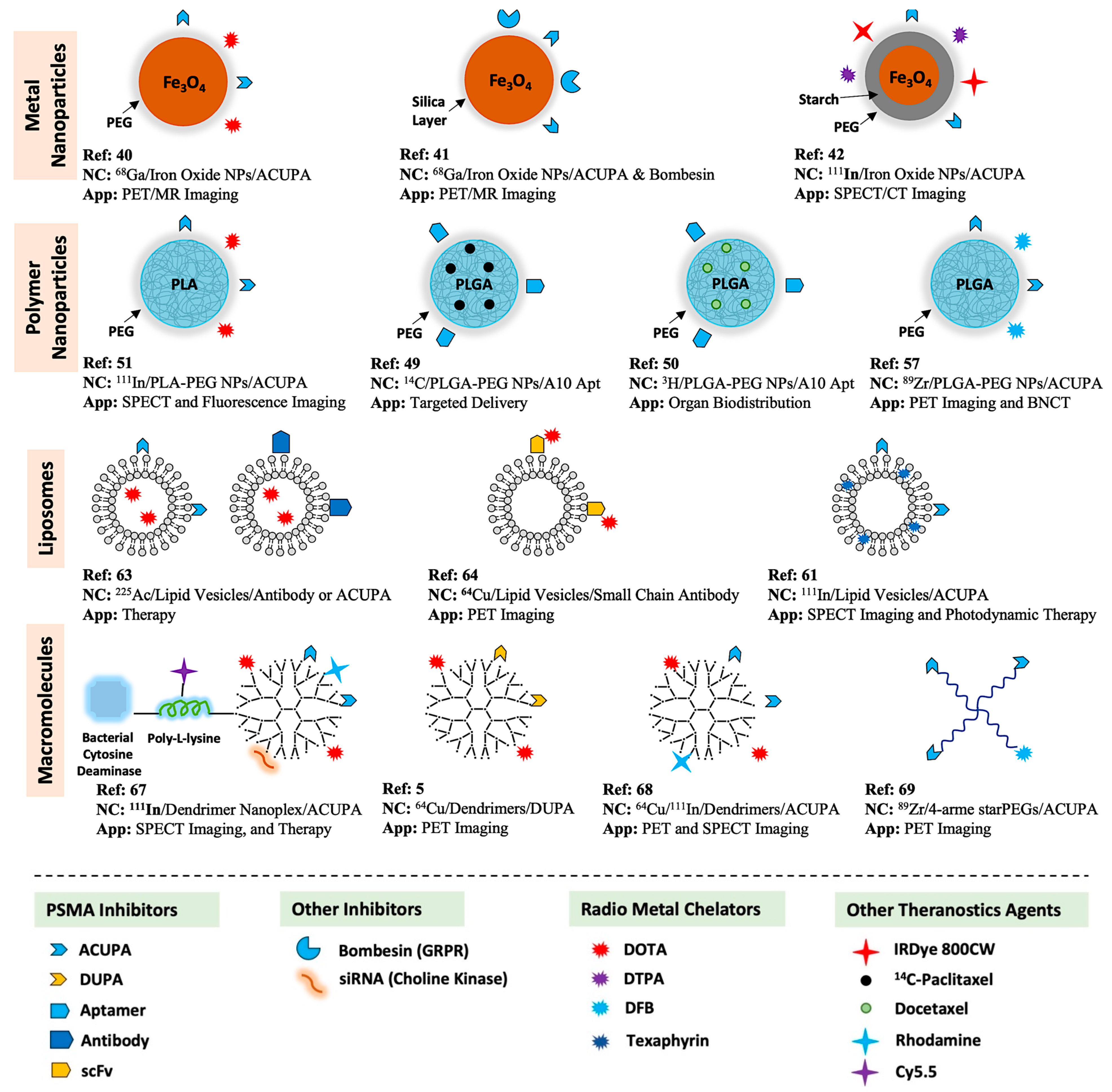
5. PSMA-Targeted Nanocarriers
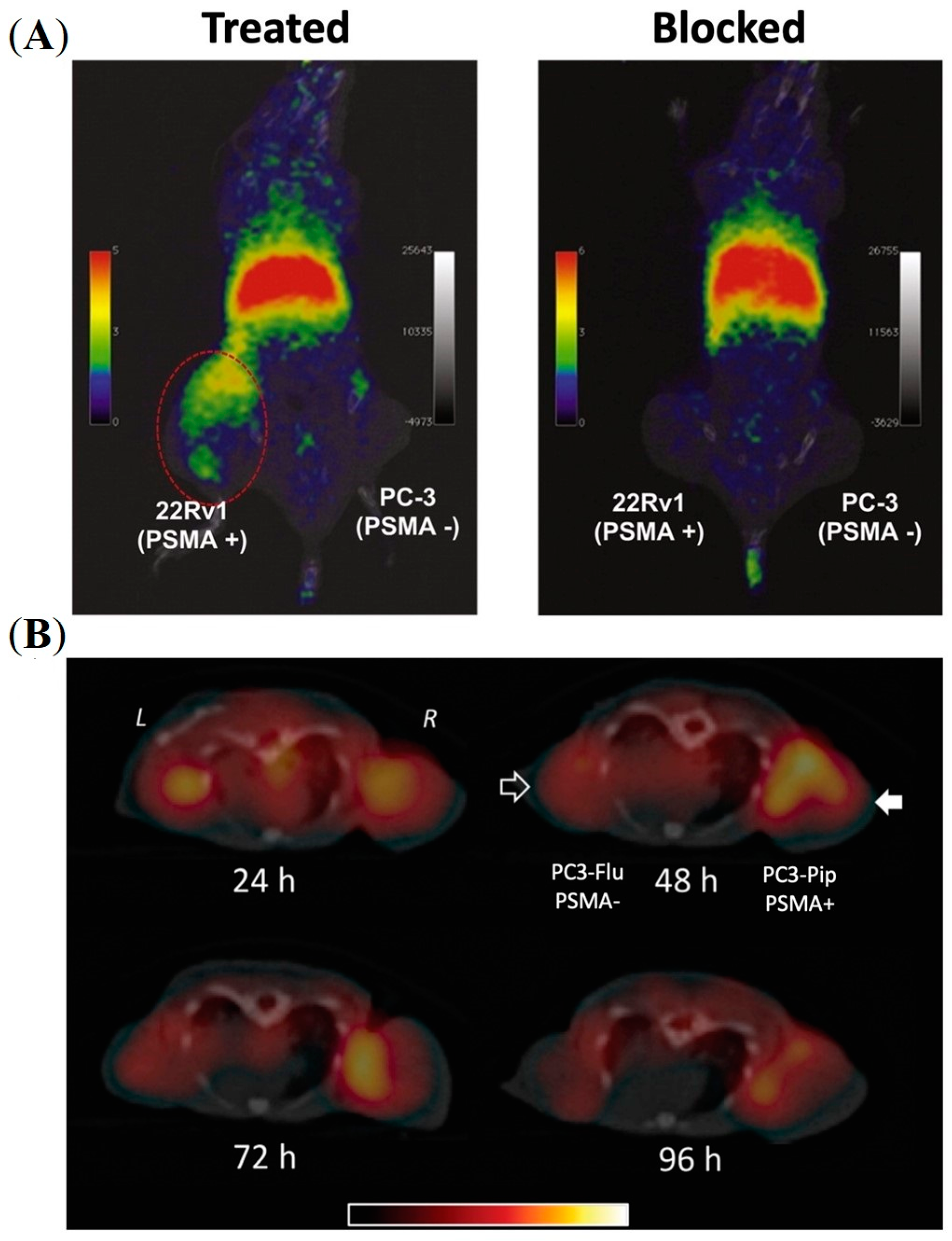
5.1. PSMA-Targeted Metal NPs

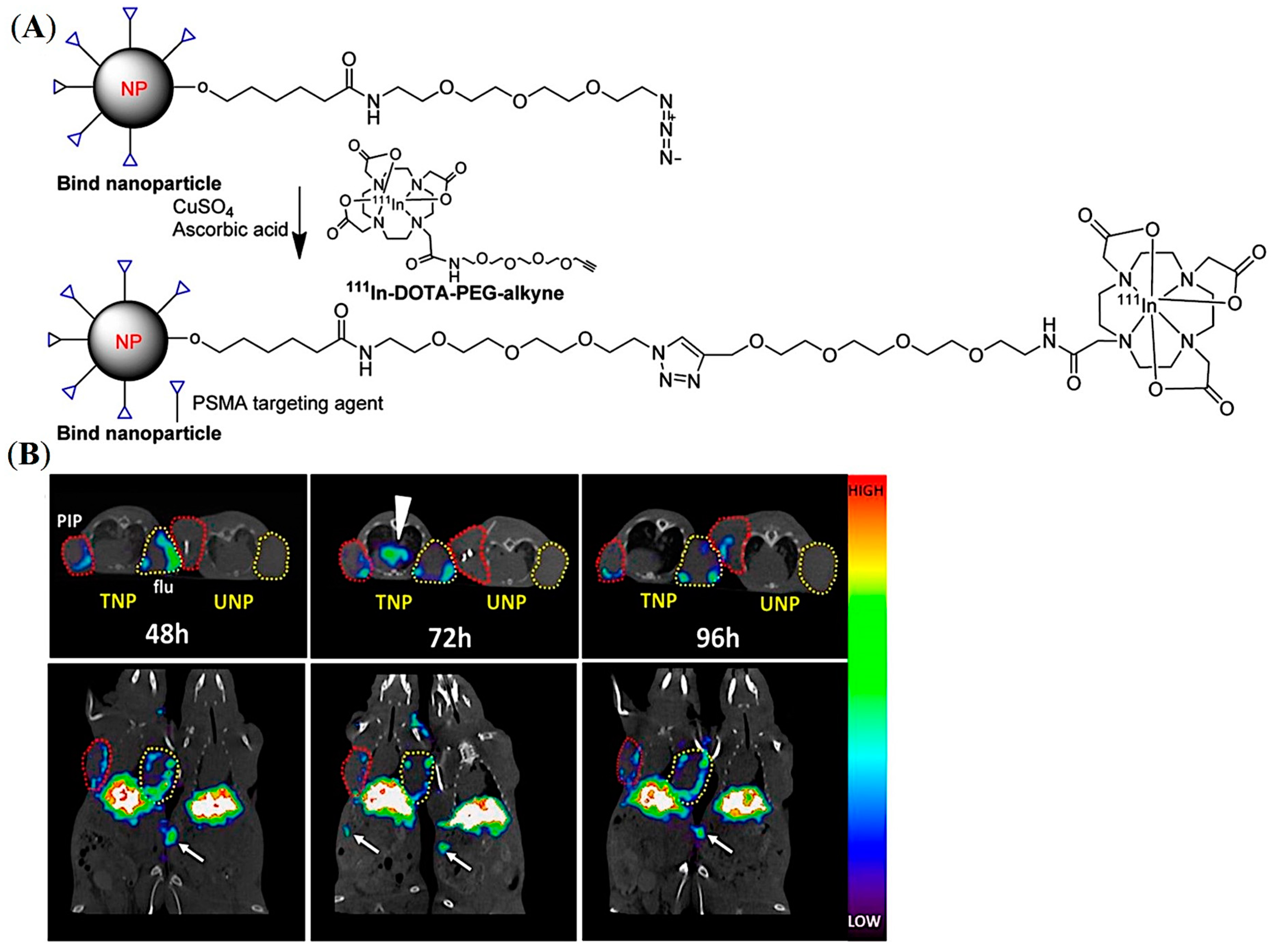
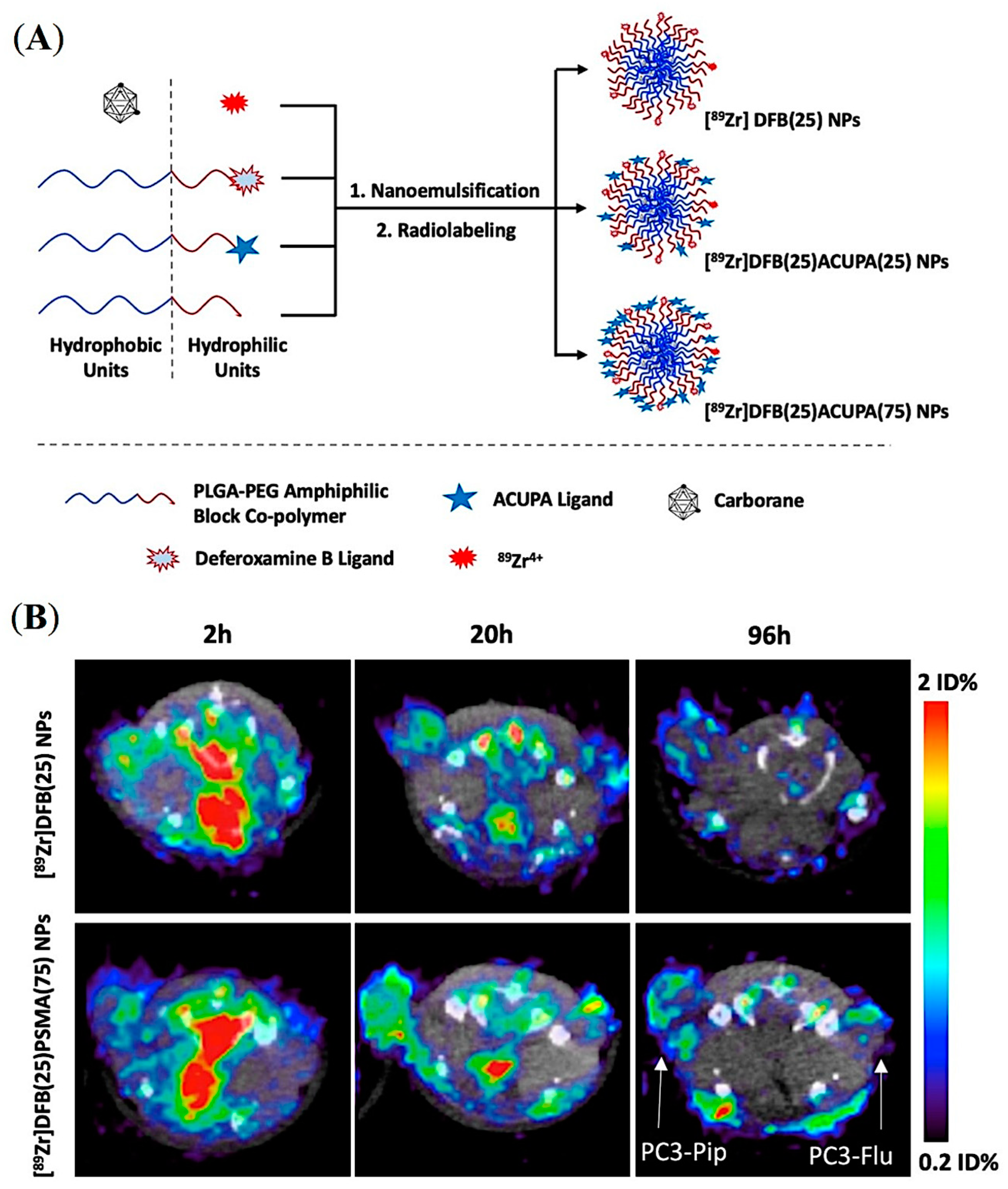
5.2. PSMA-Targeted Amphiphilic Block Copolymers
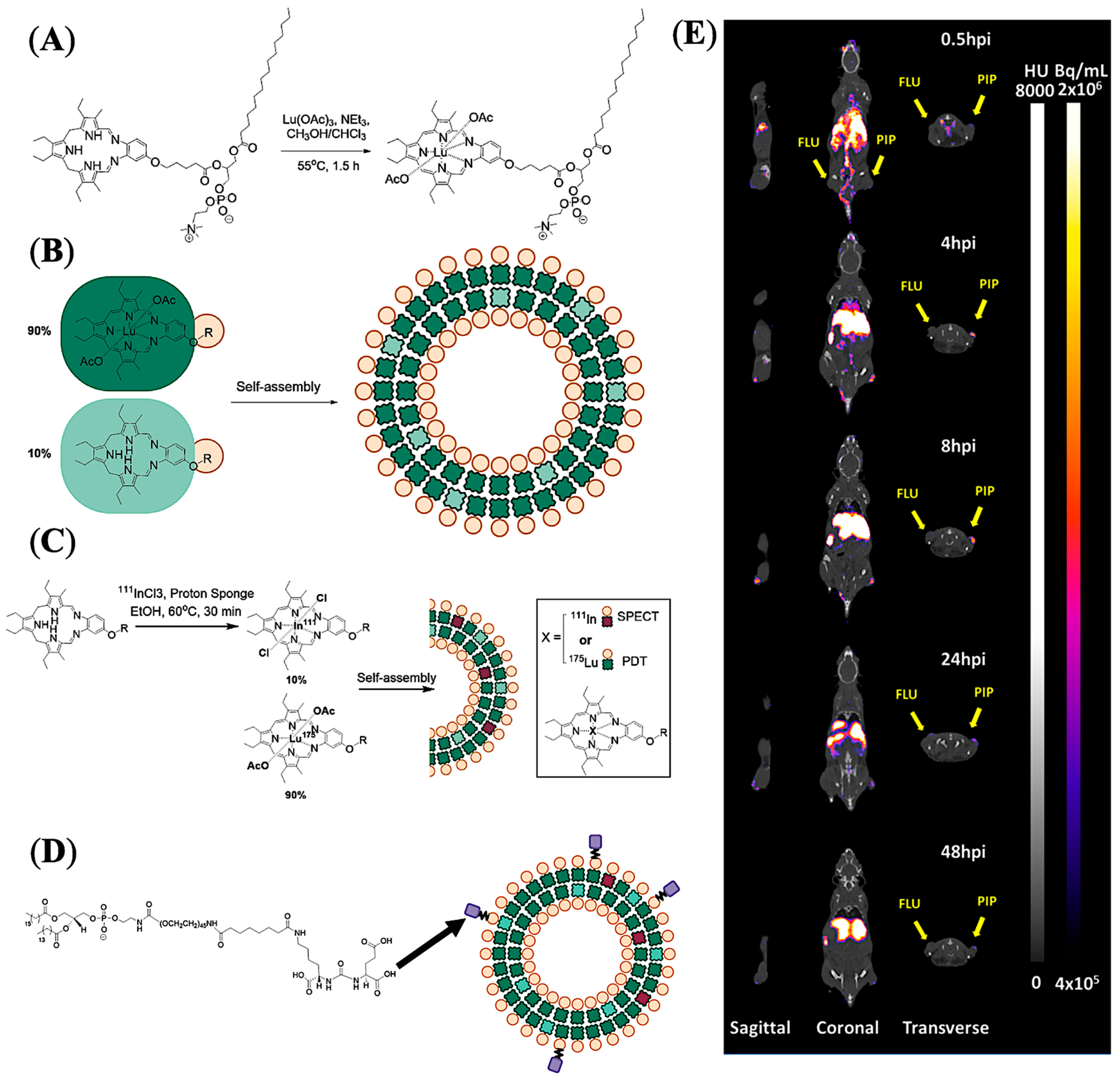
5.3. PSMA-Targeted Liposomes
5.4. PSMA-Targeted Nanoplex
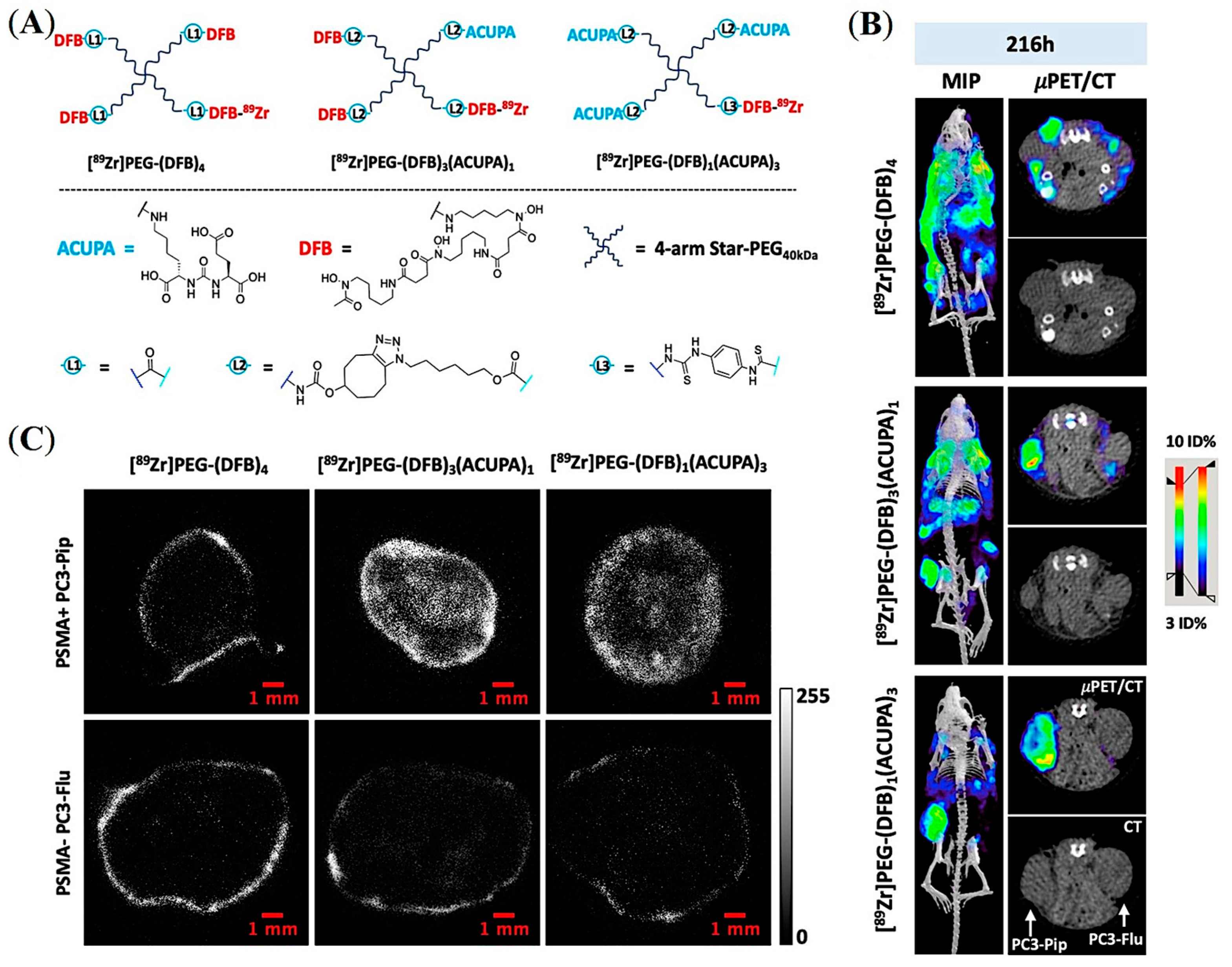
5.5. PSMA-Targeted Multivalent Dendrimers and starPEG Nanocarriers
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACUPA | ((S)-2-(3-((S)-5-amino-1- carboxypentyl) ureido) pentanedioic acid |
| BNCT | boron neutron capture therapy |
| BSB | binding site barrier |
| Chk | choline kinase |
| DFB | deferoxamine B |
| DUPA | 2-[3-(1,3-dicarboxypropyl)-ureido]pentanedioic acid |
| EPR | enhanced permeability and retention |
| FDA | Food and Drug Administration |
| GCPII | glutamate carboxypeptidase II |
| GRPR | gastrin-releasing peptide receptors |
| NPs | nanoparticles |
| PCa | prostate cancer |
| PEI | polyethyleneimine |
| PEG | polyethylene glycol |
| PET | positron emission tomography |
| PLA | polylactic acid |
| PLGA | poly(D,L-lactide-co-glycolide) |
| PSMA | prostate specific membrane antigen |
| SPECT | single-photon emission computerized tomography |
| TFA | trifluoroacetic acid |
References
- Mitchell, M.; Billingsley, M.; Haley, R.; Wechsler, M.; Peppas, N.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.; Hojjat-Farsangi, M.; Mohammadi, H.; Anvari, E.; Ghalamfarsa, G.; Yousefi, M.; Jadidi-Niaragh, F. Nanoparticles and targeted drug delivery in cancer therapy. Immunol. Lett. 2017, 190, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Senter, P.; Steinhagen, H. Drug Delivery in Oncology: From Basic Research to Cancer Therapy, Vols 1–3; Wiley: Hoboken, NJ, USA, 2012; pp. 1–1689. [Google Scholar]
- Chauhan, V.; Stylianopoulos, T.; Martin, J.; Popovic, Z.; Chen, O.; Kamoun, W.; Bawendi, M.; Fukumura, D.; Jain, R. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [Green Version]
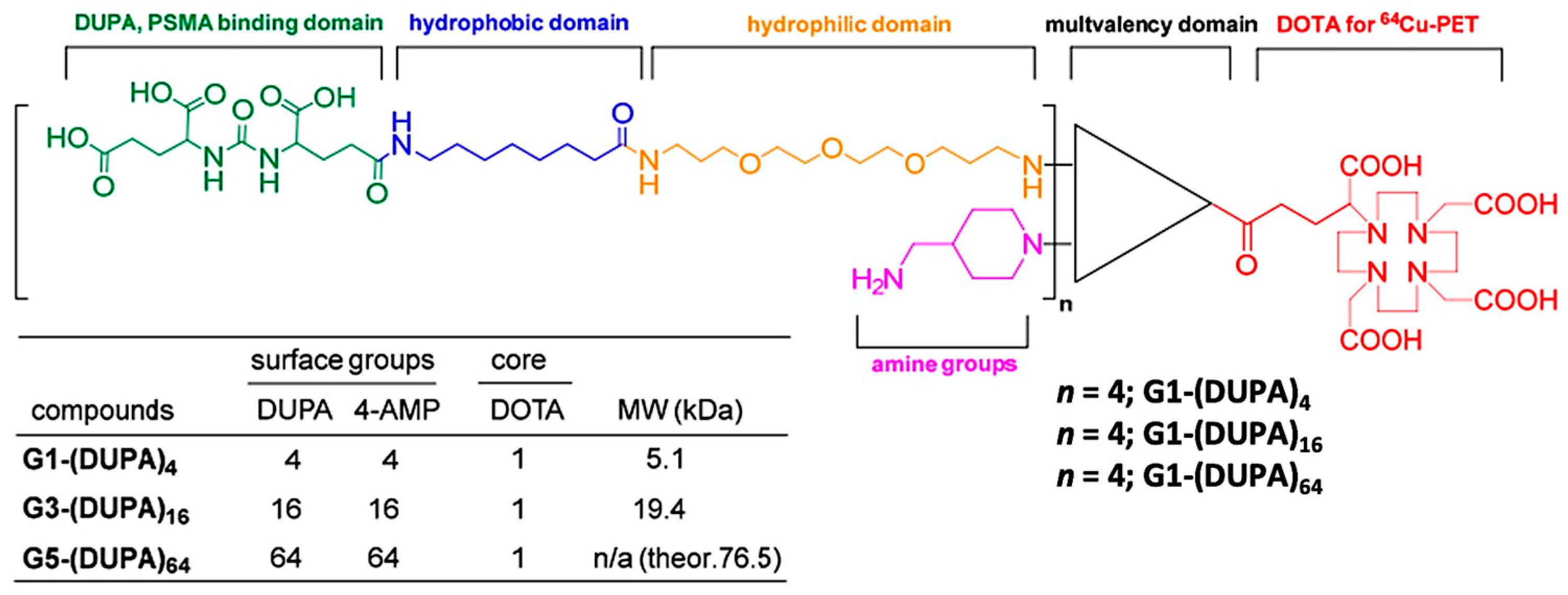
- Lim, J.; Guan, B.; Nham, K.; Hao, G.; Sun, X.; Simanek, E. Tumor Uptake of Triazine Dendrimers Decorated with Four, Sixteen, and Sixty-Four PSMA-Targeted Ligands: Passive versus Active Tumor Targeting. Biomolecules 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goos, J.; Cho, A.; Carter, L.; Dilling, T.; Davydova, M.; Mandleywala, K.; Puttick, S.; Gupta, A.; Price, W.; Quinn, J.; et al. Delivery of polymeric nanostars for molecular imaging and endoradiotherapy through the enhanced permeability and retention (EPR) effect. Theranostics 2020, 10, 567–584. [Google Scholar] [CrossRef]
- Heneweer, C.; Holland, J.; Divilov, V.; Carlin, S.; Lewis, J. Magnitude of Enhanced Permeability and Retention Effect in Tumors with Different Phenotypes: Zr-89-Albumin as a Model System. J. Nucl. Med. 2011, 52, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; He, Z.; Liu, X.; Huang, Y.; Hou, J.; Zhang, W.; Ding, D. Advances in Prostate-Specific Membrane Antigen (PSMA)-Targeted Phototheranostics of Prostate Cancer. Small Struct. 2022, 3, 2200036. [Google Scholar] [CrossRef]
- Zhou, J.; Neale, J.; Pomper, M.; Kozikowski, A. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat. Rev. Drug Discov. 2005, 4, 1015–1026. [Google Scholar] [CrossRef]
- Carter, R.; Feldman, A.; Coyle, J. Prostate-specific membrane antigen is a hydrolase with substrate and pharmacologic characteristics of a neuropeptidase. Proc. Natl. Acad. Sci. USA 1996, 93, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Bennett, M.; Thomas, L.; Bjorkman, P. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [Green Version]
- Filippi, L.; Bagni, O.; Nervi, C. Aptamer-based technology for radionuclide targeted imaging and therapy: A promising weapon against cancer. Expert Rev. Med. Devices 2020, 17, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Sah, B.; Burger, I.; Schibli, R.; Friebe, M.; Dinkelborg, L.; Graham, K.; Borkowski, S.; Bacher-Stier, C.; Valencia, R.; Srinivasan, A.; et al. Dosimetry and First Clinical Evaluation of the New F-18-Radiolabeled Bombesin Analogue BAY 864367 in Patients with Prostate Cancer. J. Nucl. Med. 2015, 56, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.; Zettlitz, K.; Tavare, R.; Kobayashi, N.; Reiter, R.; Wu, A. Dual-Modality ImmunoPET/Fluorescence Imaging of Prostate Cancer with an Anti-PSCA Cys-Minibody. Theranostics 2018, 8, 5903–5914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Chopra, S.; Trepka, K.; Wang, Y.; Sakhamuri, S.; Hooshdaran, N.; Kim, H.; Zhou, J.; Lim, S.; Leung, K.; et al. CUB Domain-Containing Protein 1 (CDCP1) Is a Target for Radioligand Therapy in Castration-Resistant Prostate Cancer, including PSMA Null Disease. Clin. Cancer Res. 2022, 28, 3066–3075. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Hua, J.; Su, Y.; Beckford-Vera, D.; Zhao, W.; Jayaraman, M.; Huynh, T.; Zhao, N.; Wang, Y.; et al. Molecular Imaging of Prostate Cancer Targeting CD46 Using ImmunoPET. Clin. Cancer Res. 2021, 27, 1305–1315. [Google Scholar] [CrossRef]
- Timmermand, O.; Elgqvist, J.; Beattie, K.; Orbom, A.; Larsson, E.; Eriksson, S.; Thorek, D.; Beattie, B.; Tran, T.; Ulmert, D.; et al. Preclinical efficacy of hK2 targeted [Lu-177] hu11B6 for prostate cancer theranostics. Theranostics 2019, 9, 2129–2142. [Google Scholar] [CrossRef]
- Korsen, J.; Kalidindi, T.; Khitrov, S.; Samuels, Z.; Chakraborty, G.; Gutierrez, J.; Poirier, J.; Rudin, C.; Chen, Y.; Morris, M.; et al. Molecular Imaging of Neuroendocrine Prostate Cancer by Targeting Delta-Like Ligand 3. J. Nucl. Med. 2022, 63, 1401–1407. [Google Scholar] [CrossRef]
- Korsen, J.; Gutierrez, J.; Tully, K.; Carter, L.; Samuels, Z.; Khitrov, S.; Poirier, J.; Rudin, C.; Chen, Y.; Morris, M.; et al. Delta-like ligand 3-targeted radioimmunotherapy for neuroendocrine prostate cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2203820119. [Google Scholar] [CrossRef]
- Chou, J.; Egusa, E.A.; Wang, S.; Badura, M.L.; Lee, F.; Bidkar, A.P.; Zhu, J.; Shenoy, T.; Trepka, K.; Robinson, T.M.; et al. Immunotherapeutic Targeting and PET Imaging of DLL3 in Small-Cell Neuroendocrine Prostate Cancer. Cancer Res. 2023, 83, 301–315. [Google Scholar] [CrossRef]
- van Rij, C.; Frielink, C.; Goldenberg, D.; Sharkey, R.; Franssen, G.; Lutje, S.; McBride, W.; Oyen, W.; Boerman, O. Pretargeted ImmunoPET of Prostate Cancer with an Anti-TROP-2 x Anti-HSG Bispecific Antibody in Mice with PC3 Xenografts. Mol. Imaging Biol. 2015, 17, 94–101. [Google Scholar] [CrossRef]
- Filippi, L.; Evangelista, L.; Sathekge, M.; Schillaci, O. ImmunoPET for prostate cancer in the PSMA era: Do we need other targets? Clin. Transl. Imaging 2022, 10, 587–596. [Google Scholar] [CrossRef]
- Virgolini, I.; Decristoforo, C.; Haug, A.; Fanti, S.; Uprimny, C. Current status of theranostics in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 471–495. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, S.; Riondato, M.; Uccelli, L.; Giovacchini, G.; Giovannini, E.; Duce, V.; Ciarmiello, A. Toward the Discovery and Development of PSMA Targeted Inhibitors for Nuclear Medicine Applications. Curr. Radiopharm. 2020, 13, 63–79. [Google Scholar] [CrossRef]
- Fendler, W.; Calais, J.; Eiber, M.; Flavell, R.; Mishoe, A.; Feng, F.; Nguyen, H.; Reiter, R.; Rettig, M.; Okamoto, S.; et al. Assessment of Ga-68-PSMA-11 PET Accuracy in Localizing Recurrent Prostate Cancer: A Prospective Single-Arm Clinical Trial. Jama Oncol. 2019, 5, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Rowe, S.; Gorin, M.; Saperstein, L.; Pouliot, F.; Josephson, D.; Wong, J.; Pantel, A.; Cho, S.; Gage, K.; et al. Diagnostic Performance of F-18-DCFPyL-PET/CT in Men with Biochemically Recurrent Prostate Cancer: Results from the CONDOR Phase III, Multicenter Study. Clin. Cancer Res. 2021, 27, 3674–3682. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.; Nordquist, L.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Cardinale, J.; Aulsebrook, M.; Gasser, G.; Mindt, T. An Overview of PET Radiochemistry, Part 2: Radiometals. J. Nucl. Med. 2018, 59, 1500–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, E.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Crisan, G.; Moldovean-Cioroianu, N.; Timaru, D.; Andries, G.; Cainap, C.; Chis, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. FDA Approves Pluvicto/Locametz for Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2022, 63, 13N. [Google Scholar]
- Filippi, L.; Chiaravalloti, A.; Schillaci, O.; Bagni, O. The potential of PSMA-targeted alpha therapy in the management of prostate cancer. Expert Rev. Anticancer Ther. 2020, 20, 823–829. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer-Chemotherapy—Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Vakoc, B.; Lanning, R.; Tyrrell, J.; Padera, T.; Bartlett, L.; Stylianopoulos, T.; Munn, L.; Tearney, G.; Fukumura, D.; Jain, R.; et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 2009, 15, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Newby, J.; Lin, C.; Zhang, L.; Xu, F.; Kim, W.; Forest, M.; Lai, S.; Milowsky, M.; Wobker, S.; et al. The Binding Site Barrier Elicited by Tumor Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. Acs Nano 2016, 10, 9243–9258. [Google Scholar] [CrossRef] [PubMed]
- Regmi, S.; Sathianathen, N.; Stout, T.; Konety, B. MRI/PET Imaging in elevated PSA and localized prostate cancer: A narrative review. Transl. Androl. Urol. 2021, 10, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.; Lawrentschuk, N.; Francis, R.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.; Frydenberg, M.; Shakher, R.; et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): A prospective, randomised, multicentre study. Lancet 2020, 395, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Yang, B.; Kim, Y.; Hong, M.; Lee, Y.; Lee, D.; Chung, J.; Jeong, J. Development of a complementary PET/MR dual-modal imaging probe for targeting prostate-specific membrane antigen (PSMA). Nanomed.-Nanotechnol. Biol. Med. 2016, 12, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Liolios, C.; Koutsikou, T.; Salvanou, E.; Kapiris, F.; Machairas, E.; Stampolaki, M.; Kolocouris, A.; Efthimiadou, E.; Bouziotis, P. Synthesis and in vitro proof-of-concept studies on bispecific iron oxide magnetic nanoparticles targeting PSMA and GRP receptors for PET/MR imaging of prostate cancer. Int. J. Pharm. 2022, 624, 122008. [Google Scholar] [CrossRef]
- Azad, B.; Banerjee, S.; Pullambhatla, M.; Lacerda, S.; Foss, C.; Wang, Y.; Ivkov, R.; Pomper, M. Evaluation of a PSMA-targeted BNF nanoparticle construct. Nanoscale 2015, 7, 4432–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ancira-Cortez, A.; Ferro-Flores, G.; Jimenez-Mancilla, N.; Morales-Avila, E.; Trujillo-Benitez, D.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Escudero-Castellanos, A.; Luna-Gutierrez, M. Synthesis, chemical and biochemical characterization of Lu2O3-iPSMA nanoparticles activated by neutron irradiation. Mater. Sci. Eng. C-Mater. Biol. Appl. 2020, 117, 111335. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Jimenez, T.; Cruz-Nova, P.; Ancira-Cortez, A.; Gibbens-Bandala, B.; Lara-Almazan, N.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Morales-Avila, E.; Ferro-Flores, G. Toxicity Assessment of [Lu-177] Lu-iFAP/iPSMA Nanoparticles Prepared under GMP-Compliant Radiopharmaceutical Processes. Nanomaterials 2022, 12, 4181. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, M.; Fracasso, G.; Pruszynski, M.; Bilewicz, A.; Kruszewski, M.; Majkowska-Pilip, A.; Lankoff, A. Design and Evaluation of 223Ra-Labeled and Anti-PSMA Targeted NaA Nanozeolites for Prostate Cancer Therapy-Part I. Materials 2020, 13, 3875. [Google Scholar] [CrossRef]
- Lankoff, A.; Czerwinska, M.; Walczak, R.; Karczmarczyk, U.; Tomczyk, K.; Brzoska, K.; Fracasso, G.; Garnuszek, P.; Mikolajczak, R.; Kruszewski, M. Design and Evaluation of Ra-223-Labeled and Anti-PSMA Targeted NaA Nanozeolites for Prostate Cancer Therapy-Part II. Toxicity, Pharmacokinetics and Biodistribution. Int. J. Mol. Sci. 2021, 22, 5702. [Google Scholar] [CrossRef]
- Hrkach, J.; Von Hoff, D.; Ali, M.; Andrianova, E.; Auer, J.; Campbell, T.; De Witt, D.; Figa, M.; Figueiredo, M.; Horhota, A.; et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012, 4, 128–139. [Google Scholar] [CrossRef]
- Afsharzadeh, M.; Hashemi, M.; Babaei, M.; Abnous, K.; Ramezani, M. PEG-PLA nanoparticles decorated with small-molecule PSMA ligand for targeted delivery of galbanic acid and docetaxel to prostate cancer cells. J. Cell. Physiol. 2020, 235, 4618–4630. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.; Levy-Nissenbaum, E.; Radovic-Moreno, A.; Langer, R.; Farokhzad, O. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Zhang, L.; Teply, B.; Mann, N.; Wang, A.; Radovic-Moreno, A.; Langer, R.; Farokhzad, O. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Foss, C.; Horhota, A.; Pullambhatla, M.; McDonnell, K.; Zale, S.; Pomper, M. In-111- and IRDye800CW-Labeled PLA-PEG Nanoparticle for Imaging Prostate-Specific Membrane Antigen-Expressing Tissues. Biomacromolecules 2017, 18, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.; Zhang, M. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Barth, R.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, S.; Vicente, M.d.G.H. Recent Advances in Boron Delivery Agents for Boron Neutron Capture Therapy (BNCT). In Boron-Based Compounds; Wiley: Hoboken, NJ, USA, 2018; pp. 298–342. [Google Scholar]
- Haapaniemi, A.; Kankaanranta, L.; Saat, R.; Koivunoro, H.; Saarilahti, K.; Makitie, A.; Atula, T.; Joensuu, H. Boron Neutron Capture Therapy in the Treatment of Recurrent Laryngeal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Blaha, C.; Santos, R.; Huynh, T.; Hayes, T.; Beckford-Vera, D.; Blecha, J.; Hong, A.; Fogarty, M.; Hope, T.; et al. Synthesis and Initial Biological Evaluation of Boron-Containing Prostate-Specific Membrane Antigen Ligands for Treatment of Prostate Cancer Using Boron Neutron Capture Therapy. Mol. Pharm. 2019, 16, 3831–3841. [Google Scholar] [CrossRef]
- Meher, N.; Seo, K.; Wang, S.; Bidkar, A.; Fogarty, M.; Dhrona, S.; Huang, X.; Tang, R.; Blaha, C.; Evans, M.; et al. Synthesis and Preliminary Biological Assessment of Carborane-Loaded Theranostic Nanoparticles to Target Prostate-Specific Membrane Antigen. Acs Appl. Mater. Interfaces 2021, 13, 54739–54752. [Google Scholar] [CrossRef]
- Vera, D.; Fontaine, S.; VanBrocklin, H.; Hearn, B.; Reid, R.; Ashley, G.; Santi, D. PET Imaging of the EPR Effect in Tumor Xenografts Using Small 15 nm Diameter Polyethylene Glycols Labeled with Zirconium-89. Mol. Cancer Ther. 2020, 19, 673–679. [Google Scholar] [CrossRef]
- Chen, Z.; Tai, Z.; Gu, F.; Hu, C.; Zhu, Q.; Gao, S. Aptamer-mediated delivery of docetaxel to prostate cancer through polymeric nanoparticles for enhancement of antitumor efficacy. Eur. J. Pharm. Biopharm. 2016, 107, 130–141. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.; Sood, A.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Overchuk, M.; Rajora, M.; Lou, J.; Chen, Y.; Pomper, M.; Chen, J.; Zheng, G. Targeted Theranostic 111In/Lu-Nanotexaphyrin for SPECT Imaging and Photodynamic Therapy. Mol. Pharm. 2022, 19, 1803–1813. [Google Scholar] [CrossRef]
- Vaughan, L.; Glanzel, W.; Korch, C.; Capes-Davis, A. Widespread Use of Misidentified Cell Line KB (HeLa): Incorrect Attribution and Its Impact Revealed through Mining the Scientific Literature. Cancer Res. 2017, 77, 2784–2788. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Bandekar, A.; Sempkowski, M.; Banerjee, S.; Pomper, M.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Nanoconjugation of PSMA-Targeting Ligands Enhances Perinuclear Localization and Improves Efficacy of Delivered Alpha-Particle Emitters against Tumor Endothelial Analogues. Mol. Cancer Ther. 2016, 15, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
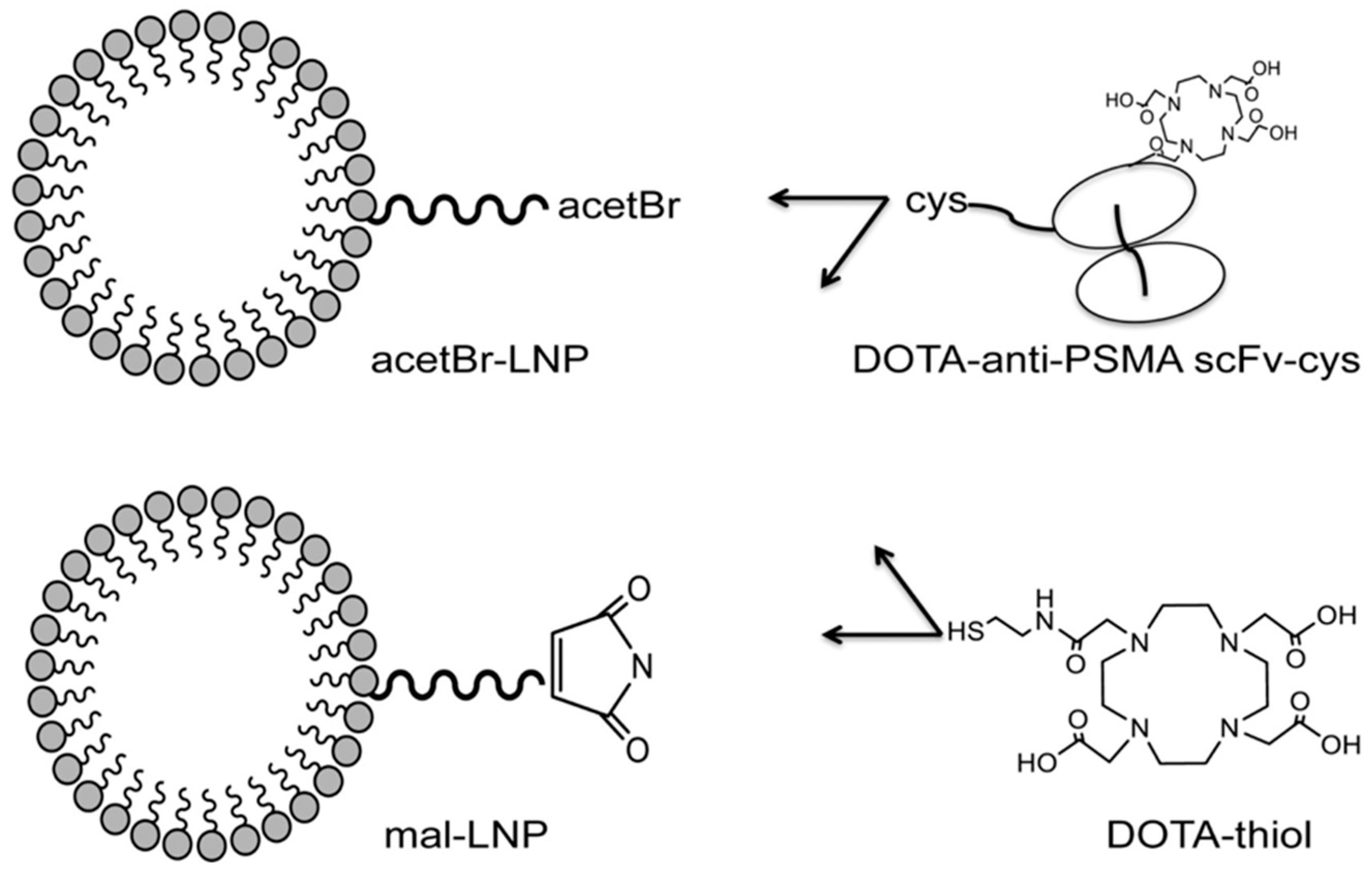
- Wong, P.; Li, L.; Chea, J.; Delgado, M.; Crow, D.; Poku, E.; Szpikowska, B.; Bowles, N.; Channappa, D.; Colcher, D.; et al. PET imaging of Cu-64-DOTA-scFv-anti-PSMA lipid nanoparticles (LNPs): Enhanced tumor targeting over anti-PSMA scFv or untargeted LNPs. Nucl. Med. Biol. 2017, 47, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.; Li, L.; Chea, J.; Delgado, M.; Poku, E.; Szpikowska, B.; Bowles, N.; Minnix, M.; Colcher, D.; Wong, J.; et al. Synthesis, Positron Emission Tomography Imaging, and Therapy of Diabody Targeted Drug Lipid Nanoparticles in a Prostate Cancer Murine Model. Cancer Biother. Radiopharm. 2017, 32, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Zuckerman, J.; Choi, C.; Seligson, D.; Tolcher, A.; Alabi, C.; Yen, Y.; Heidel, J.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [Green Version]
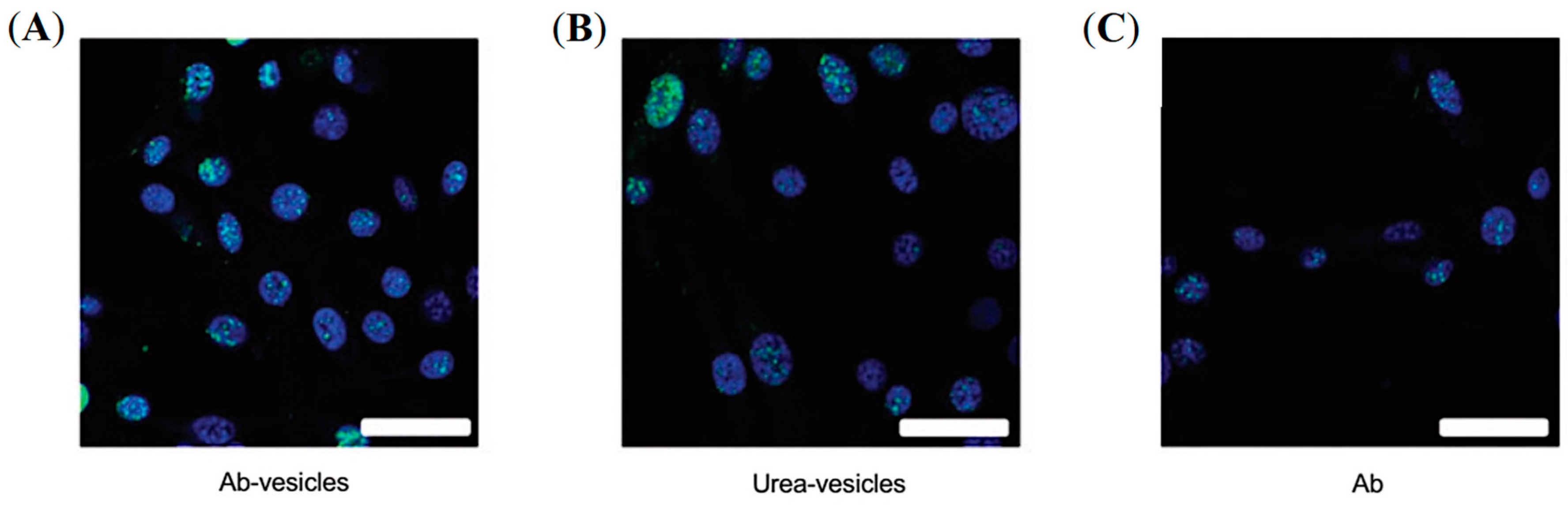
- Chen, Z.; Penet, M.; Nimmagadda, S.; Li, C.; Banerjee, S.; Winnard, P.; Artemov, D.; Glunde, K.; Pomper, M.; Bhujwalla, Z. PSMA-Targeted Theranostic Nanoplex for Prostate Cancer Therapy. Acs Nano 2012, 6, 7752–7762. [Google Scholar] [CrossRef] [Green Version]
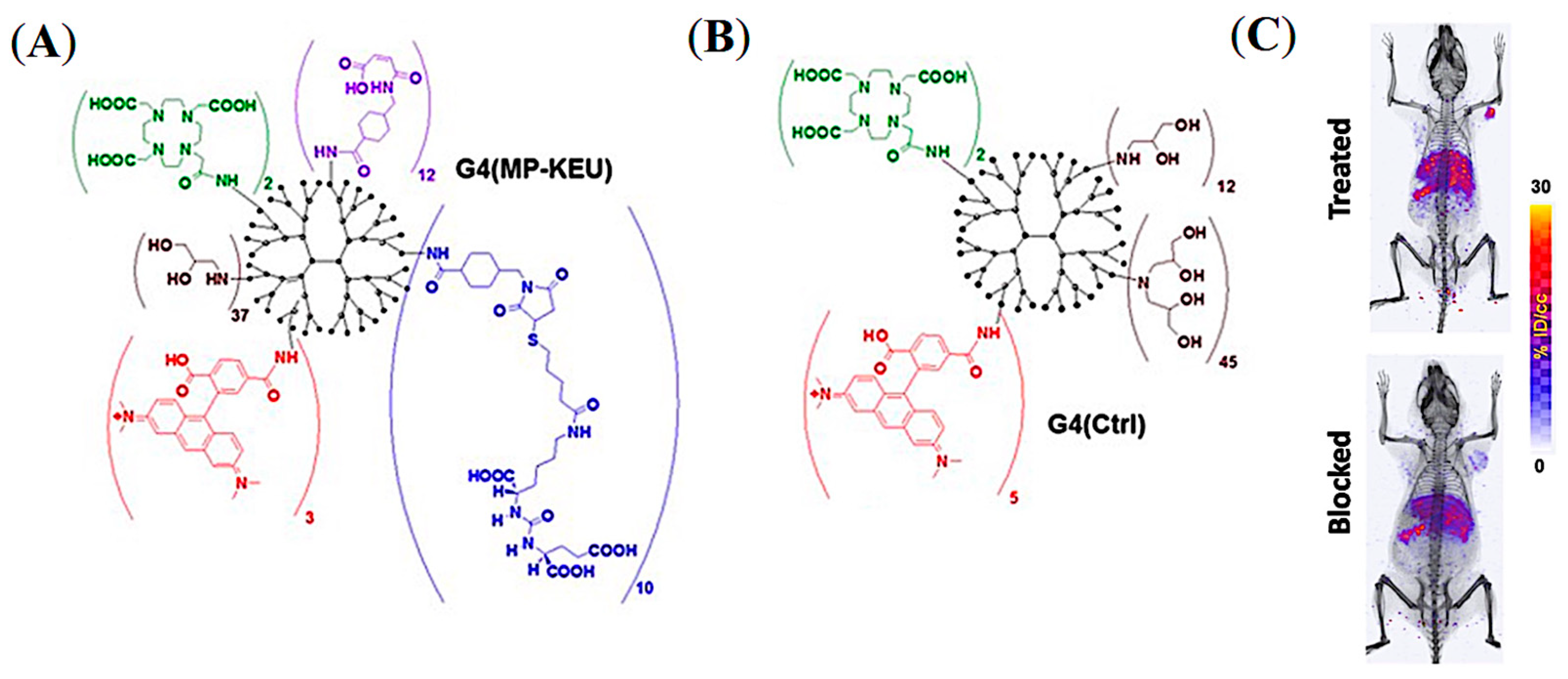
- Lesniak, W.; Boinapally, S.; Banerjee, S.; Azad, B.; Foss, C.; Shen, C.; Lisok, A.; Wharram, B.; Nimmagadda, S.; Pomper, M. Evaluation of PSMA-Targeted PAMAM Dendrimer Nanoparticles in a Murine Model of Prostate Cancer. Mol. Pharm. 2019, 16, 2590–2604. [Google Scholar] [CrossRef]
- Meher, N.; Ashley, G.; Bidkar, A.; Dhrona, S.; Fong, C.; Fontaine, S.; Vera, D.; Wilson, D.; Seo, Y.; Santi, D.; et al. Prostate-Specific Membrane Antigen Targeted Deep Tumor Penetration of Polymer Nanocarriers. Acs Appl. Mater. Interfaces 2022, 14, 50569–50582. [Google Scholar] [CrossRef]
- Gratton, S.; Ropp, P.; Pohlhaus, P.; Luft, J.; Madden, V.; Napier, M.; DeSimone, J. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Safari, H.; Kelley, W.; Saito, E.; Kaczorowski, N.; Carethers, L.; Shea, L.; Eniola-Adefeso, O. Neutrophils preferentially phagocytose elongated particles-An opportunity for selective targeting in acute inflammatory diseases. Sci. Adv. 2020, 6, eaba1474. [Google Scholar] [CrossRef]
- Elci, S.; Jiang, Y.; Yan, B.; Kim, S.; Saha, K.; Moyano, D.; Tonga, G.; Jackson, L.; Rotello, V.; Vachet, R. Surface Charge Controls the Suborgan Biodistributions of Gold Nanoparticles. Acs Nano 2016, 10, 5536–5542. [Google Scholar] [CrossRef]
- Pijeira, M.; Viltres, H.; Kozempel, J.; Sakmar, M.; Vlk, M.; Ilem-Ozdemir, D.; Ekinci, M.; Srinivasan, S.; Rajabzadeh, A.; Ricci-Junior, E.; et al. Radiolabeled nanomaterials for biomedical applications: Radiopharmacy in the era of nanotechnology. Ejnmmi Radiopharm. Chem. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Androvic, L.; Woldrichova, L.; Jozefjakova, K.; Pechar, M.; Lynn, G.; Kankova, D.; Malinova, L.; Laga, R. Cyclotriphosphazene-Based Star Copolymers as Structurally Tunable Nanocarriers with Programmable Biodegradability. Macromolecules 2021, 54, 3139–3157. [Google Scholar] [CrossRef]
- Ernsting, M.; Murakami, M.; Roy, A.; Li, S. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.; Maecke, H.; Borner, A.; Weckesser, E.; Schoffski, P.; Oei, M.; Schumacher, J.; Henze, M.; Heppeler, A.; Meyer, G.; et al. Biokinetics and imaging with the somatostatin receptor PET radioligand Ga-68-DOTATOC: Preliminary data. Eur. J. Nucl. Med. 2001, 28, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Debela, D.; Muzazu, S.; Heraro, K.; Ndalama, M.; Mesele, B.; Haile, D.; Kitui, S.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. Sage Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Structures | CA | CN | Radiometals |
|---|---|---|---|---|
| DFO; desferrioxamine B |  | O6 | 6 | 89Zr4+ |
| DTPA; diethylenetriaminepentaacetic acid |  | N3O5 | 8 | 111In3+, 177Lu3+, 86/90Y3+ |
| Pa Family; H2dedpa; 1,2-[[6-(carboxy)-pyridin-2-yl]- methylamino]ethane |  | N4O2 | 6 | 67/68Ga3+,111In3+, 177Lu3+ |
| HOPO; 3,4,3-(LI-1,2-HOPO) |  | O8 | 8 | 89Zr4+, 227Th4+ |
| NOTA; 1,4,7-triazacyclononane-1,4,7- triacetic acid |  | N3O3 | 6 | 177Lu3+, 64Cu2+, 67/68Ga3+, 86/90Y3+, 212/213Bi3+ |
| DOTA; 1,4,7,10-tetraazacyclododecane- 1,4,7,10-tetraacetic acid, maximum |  | N4O4 | 8 | 64Cu2+, 212Pb2+, 212/213Bi3+, 177Lu3+, 225Ac3+, 111In3+, 44/47Sc3+, 86/90Y3+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meher, N.; VanBrocklin, H.F.; Wilson, D.M.; Flavell, R.R. PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer. Pharmaceuticals 2023, 16, 315. https://doi.org/10.3390/ph16020315
Meher N, VanBrocklin HF, Wilson DM, Flavell RR. PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer. Pharmaceuticals. 2023; 16(2):315. https://doi.org/10.3390/ph16020315
Chicago/Turabian StyleMeher, Niranjan, Henry F. VanBrocklin, David M. Wilson, and Robert R. Flavell. 2023. "PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer" Pharmaceuticals 16, no. 2: 315. https://doi.org/10.3390/ph16020315