
Protein Interactome of Amyloid-β as a Therapeutic Target
 , and
, and 
Abstract
:1. Introduction
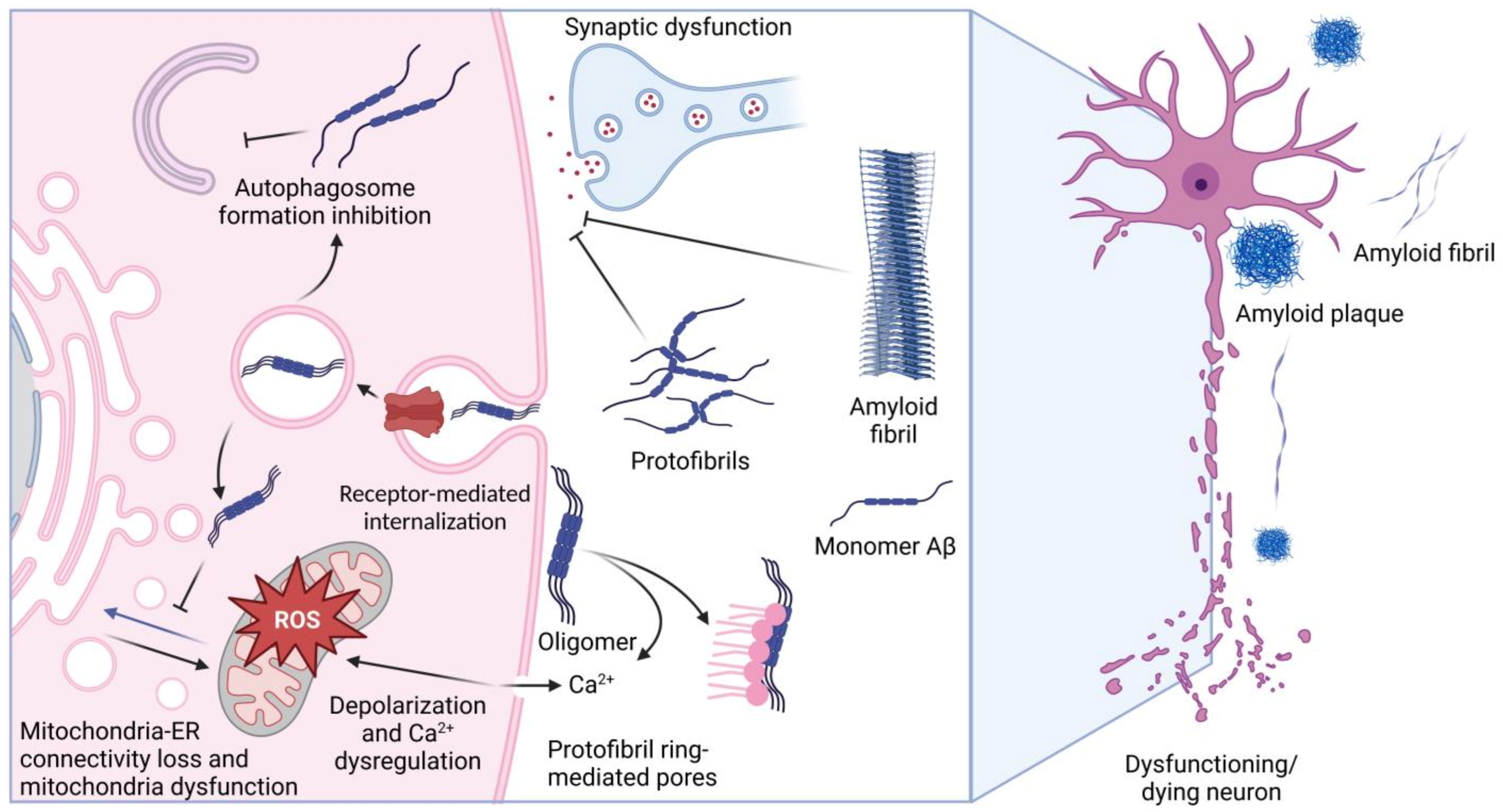
2. Aβ and Its Protein Interactors
2.1. Structural Features of Aβ
2.2. Protein Interactome of Aβ
2.3. Proteins Whose Function Is Affected by Aβ
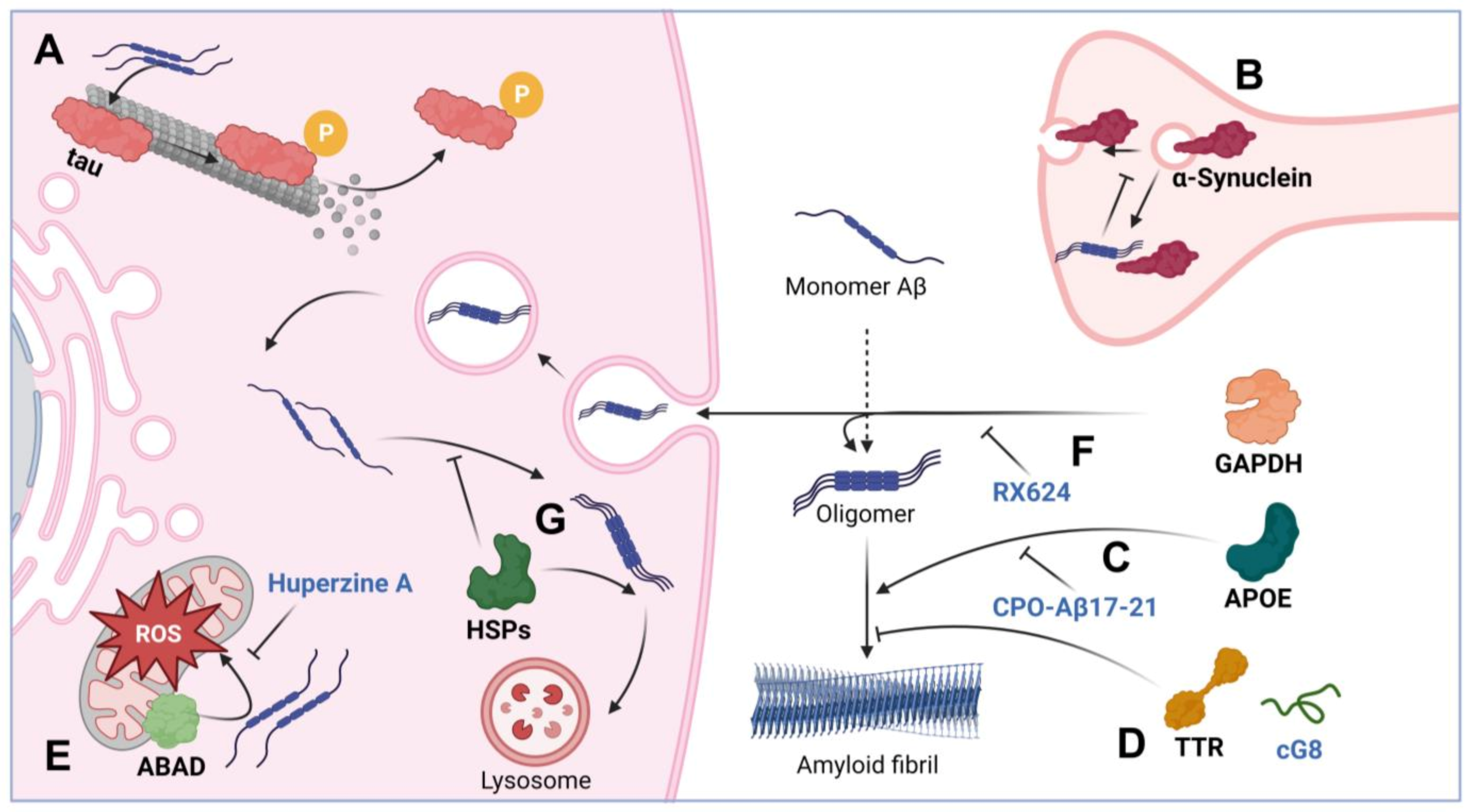
2.3.1. Tau-Protein
2.3.2. α-Synuclein
2.4. Proteins Affecting Aβ Toxicity
2.4.1. Apolipoprotein E
2.4.2. Transthyretin
2.4.3. ABAD
2.4.4. GAPDH
2.4.5. Chaperones
2.4.6. Cystatin C
3. Chemicals Targeting Aβ and Its Intermolecular Complexes

{kind=link}
{kind=link}
| Therapeutical Agent (Class of Agents) | Potential Function | Reference |
|---|---|---|
| Aducanumab (specific antibodies) and other agents preventing formation of Aβ fibrils formation | Prevention of Aβ assemblage into cytotoxic fibrils | [113] |
| Synthetic and natural peptides that may block amyloid–amyloid binding | Interaction conditioned by the similarity to the hydrophobic domains of Aβ | [117] |
| Small molecules able to inhibit Aβ misfolding and enhance its clearance (LS4, for example) | Specifically binding to different soluble forms of Aβ | [120] |
| CPO-Aβ17–21 peptide | Blocking the ability of APOE to initiate Aβ oligomerization | [122] |
| Cyclic peptide cG8 | TTR-mimetic peptide comprising its Aβ-binding domain | [124] |
| Huperzine A and other ABAD blocking compounds | ABAD inhibition reduces Aβ-induced mitochondrial dysfunction | [127] |
| GAPDH–Aβ complex inhibitors | Blocking the formation of the GAPDH–Aβ complex and reduction of its cytotoxicity | [94,138] |
| Chaperone synthesis inducers | Newly synthesized chaperones block the formation of Aβ complexes with other proteins | [131,136,137] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Cano, S.S.; Elio, I.; Vergara, M.M.; Giampieri, F.; Battino, M. Associations between Sleep, Cortisol Regulation, and Diet: Possible Implications for the Risk of Alzheimer Disease. Adv. Nutr. 2016, 7, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; De Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.B.; Wyart, C.; Buldyrev, S.V.; Cruz, L.; Urbanc, B.; Hasselmo, M.E.; Stanley, H.E.; Hyman, B.T. Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 5274–5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, G.A.; Levi, C.R.; Schofield, P.; Wang, Y.; Lovett, E.C. Quantitative measurement of cerebral haemodynamics in early vascular dementia and Alzheimer’s disease. J. Clin. Neurosci. 2006, 13, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers. Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 2021, 144, 310–324. [Google Scholar] [CrossRef]
- Wiatrak, B.; Piasny, J.; Kuźniarski, A.; Gąsiorowski, K. Interactions of Amyloid-β with Membrane Proteins. Int. J. Mol. Sci. 2021, 22, 6075. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Villaflores, O.B.; Chen, Y.J.; Chen, C.P.; Yeh, J.M.; Wu, T.Y. Curcuminoids and resveratrol as anti-Alzheimer agents. Taiwan. J. Obstet. Gynecol. 2012, 51, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar]
- Hoe, H.S.; Fu, Z.; Makarova, A.; Lee, J.Y.; Lu, C.; Feng, L.; Pajoohesh-Ganji, A.; Matsuoka, Y.; Hyman, B.T.; Ehlers, M.D.; et al. The effects of amyloid precursor protein on postsynaptic composition and activity. J. Biol. Chem. 2009, 284, 8495–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.C.; Ludewig, S.; Winschel, A.; Abel, T.; Bold, C.; Salzburger, L.R.; Klein, S.; Han, K.; Weyer, S.W.; Fritz, A.; et al. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. EMBO J. 2018, 37, e98335. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Raveh, A.; Schneider, A.; Fritzius, T.; Früh, S.; Rem, P.D.; Stawarski, M.; Lalanne, T.; Turecek, R.; Choo, M.; et al. Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat. Commun. 2019, 10, 1331. [Google Scholar] [CrossRef] [Green Version]
- Hefter, D.; Ludewig, S.; Draguhn, A.; Korte, M. Amyloid, APP, and Electrical Activity of the Brain. Neuroscientist 2020, 26, 231–251. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.K.; Pittman, J.M.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. β-Amyloid aggregation and heterogeneous nucleation. Protein Sci. 2019, 28, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [Green Version]
- Rice, H.C.; De Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 2019, 363, eaao4827. [Google Scholar] [CrossRef]
- McLaurin, J.; Lai, A.Y. Mechanisms of amyloid-Beta Peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimers. Dis. 2010, 2011, 548380. [Google Scholar]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Naia, L.; Piras, A.; Graff, C.; Cattaneo, A.; Meli, G.; Hamasaki, M.; Nilsson, P.; et al. Amyloid Β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models. Cells 2020, 9, 2552. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canevari, L.; Abramov, A.Y.; Duchen, M.R. Toxicity of amyloid beta peptide: Tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004, 29, 637–650. [Google Scholar] [CrossRef]
- Lord, A.; Englund, H.; Söderberg, L.; Tucker, S.; Clausen, F.; Hillered, L.; Gordon, M.; Morgan, D.; Lannfelt, L.; Pettersson, F.E.; et al. Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 2009, 276, 995–1006. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef] [Green Version]
- Penke, B.; Szucs, M.; Bogár, F. Oligomerization and Conformational Change Turn Monomeric β-Amyloid and Tau Proteins Toxic: Their Role in Alzheimer’s Pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef] [Green Version]
- Jamasbi, E.; Separovic, F.; Hossain, M.A.; Ciccotosto, G.D. Phosphorylation of a full length amyloid-β peptide modulates its amyloid aggregation, cell binding and neurotoxic properties. Mol. Biosyst. 2017, 13, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Mezentsev, Y.V.; Medvedev, A.E.; Kechko, O.I.; Makarov, A.A.; Ivanov, A.S.; Mantsyzov, A.B.; Kozin, S.A. Zinc-induced heterodimer formation between metal-binding domains of intact and naturally modified amyloid-beta species: Implication to amyloid seeding in Alzheimer’s disease? J. Biomol. Struct. Dyn. 2016, 34, 2317–2326. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na, K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers. Res. Ther. 2014, 6, 28–29. [Google Scholar] [CrossRef] [Green Version]
- Yurinskaya, M.M.; Mitkevich, V.A.; Kozin, S.A.; Evgenev, M.B.; Makarov, A.A.; Vinokurov, M.G. HSP70 protects human neuroblastoma cells from apoptosis and oxidative stress induced by amyloid peptide isoAsp7-Aβ(1-42). Cell Death Dis. 2015, 6, e1977. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 2013, 4, e939. [Google Scholar] [CrossRef] [Green Version]
- Bayer, T.A. Pyroglutamate Aβ cascade as drug target in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 1880. [Google Scholar] [CrossRef]
- Wirths, O.; Breyhan, H.; Cynis, H.; Schilling, S.; Demuth, H.U.; Bayer, T.A. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009, 118, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [Green Version]
- Forester, B.P.; Berlow, Y.A.; Harper, D.G.; Jensen, J.E.; Lange, N.; Froimowitz, M.P.; Ravichandran, C.; Iosifescu, D.V.; Lukas, S.E.; Renshaw, P.F.; et al. Age-related changes in brain energetics and phospholipid metabolism. NMR Biomed. 2010, 23, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Hossain, M.S.; Bilkis, T.; Islam, M.I.; Park, I.S. Evidence for a Strong Relationship between the Cytotoxicity and Intracellular Location of β-Amyloid. Life 2022, 12, 577. [Google Scholar] [CrossRef]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef] [PubMed]
- Oláh, J.; Vincze, O.; Virók, D.; Simon, D.; Bozsó, Z.; Tokési, N.; Horváth, I.; Hlavanda, E.; Kovács, J.; Magyar, A.; et al. Interactions of pathological hallmark proteins: Tubulin polymerization promoting protein/p25, beta-amyloid, and alpha-synuclein. J. Biol. Chem. 2011, 286, 34088–34100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, L.; Kakuyama, H.; Möllert, A.; Ito, A.; Winblad, B.; Tjernberg, L.O.; Näslund, J. Characterization of the Alzheimer’s disease-associated CLAC protein and identification of an amyloid beta-peptide-binding site. J. Biol. Chem. 2005, 280, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040. [Google Scholar] [CrossRef]
- Gerber, H.; Mosser, S.; Boury-Jamot, B.; Stumpe, M.; Piersigilli, A.; Goepfert, C.; Dengjel, J.; Albrecht, U.; Magara, F.; Fraering, P.C. The APMAP interactome reveals new modulators of APP processing and beta-amyloid production that are altered in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 13. [Google Scholar] [CrossRef]
- Panikker, P.; Xu, S.J.; Zhang, H.; Sarthi, J.; Beaver, M.; Sheth, A.; Akhter, S.; Elefant, F. Restoring Tip60 HAT/HDAC2 Balance in the Neurodegenerative Brain Relieves Epigenetic Transcriptional Repression and Reinstates Cognition. J. Neurosci. 2018, 38, 4569–4583. [Google Scholar] [CrossRef] [Green Version]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pichet Binette, A.; Franzmeier, N.; Spotorno, N.; Ewers, M.; Brendel, M.; Biel, D.; Weiner, M.; Aisen, P.; Petersen, R.; Jack, C.R.; et al. Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer’s disease. Nat. Commun. 2022, 13, 6635. [Google Scholar] [CrossRef] [PubMed]
- Aliakbari, F.; Attar, F.; Movahedi, M.; Falahati, M. Human tau fibrillization and neurotoxicity in the presence of magnesium oxide nanoparticle fabricated through laser ablation method. Spectrochim. Acta. Part A Mol. Biomol. Spectrosc. 2022, 278, 121372. [Google Scholar] [CrossRef] [PubMed]
- Scholz, T.; Mandelkow, E. Transport and diffusion of Tau protein in neurons. Cell Mol. Life Sci. 2014, 71, 3139–3150. [Google Scholar] [CrossRef]
- Fan, Q.W.; Yu, W.; Senda, T.; Yanagisawa, K.; Michikawa, M. Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J. Neurochem. 2001, 76, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Aragão Gomes, L.; Uytterhoeven, V.; Lopez-Sanmartin, D.; Tomé, S.O.; Tousseyn, T.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Verstreken, P.; Thal, D.R. Maturation of neuronal AD-tau pathology involves site-specific phosphorylation of cytoplasmic and synaptic tau preceding conformational change and fibril formation. Acta Neuropathol. 2021, 141, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Tomé, S.O. The central role of tau in Alzheimer’s disease: From neurofibrillary tangle maturation to the induction of cell death. Brain Res. Bull. 2022, 190, 204–217. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dubois, C.; Perez, K.; Varghese, S.; Birchall, I.E.; Leckey, M.; Davydova, N.; McLean, C.; Nisbet, R.M.; Roberts, B.R.; et al. Quantitative proteomics of tau and Aβ in detergent fractions from Alzheimer’s disease brains. J. Neurochem. 2022. [Google Scholar] [CrossRef]
- Lam, S.; Hérard, A.S.; Boluda, S.; Petit, F.; Eddarkaoui, S.; Cambon, K.; Letournel, F.; Martin-Négrier, M.L.; Faisant, M.; Godfraind, C.; et al. Pathological changes induced by Alzheimer’s brain inoculation in amyloid-beta plaque-bearing mice. Acta Neuropathol. Commun. 2022, 10, 112. [Google Scholar] [CrossRef]
- Ikezu, S.; Ingraham Dixie, K.L.; Koro, L.; Watanabe, T.; Kaibuchi, K.; Ikezu, T. Tau-tubulin kinase 1 and amyloid-β peptide induce phosphorylation of collapsin response mediator protein-2 and enhance neurite degeneration in Alzheimer disease mouse models. Acta Neuropathol. Commun. 2020, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: Implications to synaptic dysfunction and neuronal damage. J. Alzheimers. Dis. 2013, 36, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Atias, M.; Tevet, Y.; Sun, J.; Stavsky, A.; Tal, S.; Kahn, J.; Roy, S.; Gitler, D. Synapsins regulate α-synuclein functions. Proc. Natl. Acad. Sci. USA 2019, 166, 11116–11118. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.M.; Thorn, D.C.; Dobson, C.M.; Meehan, S.; Jackson, S.E.; Woodcock, J.M.; Carver, J.A. The Amyloid Fibril-Forming β-Sheet Regions of Amyloid β and α-Synuclein Preferentially Interact with the Molecular Chaperone 14-3-3ζ. Molecules 2021, 26, 6120. [Google Scholar] [CrossRef] [PubMed]
- Köppen, J.; Schulze, A.; Machner, L.; Wermann, M.; Eichentopf, R.; Guthardt, M.; Hähnel, A.; Klehm, J.; Kriegeskorte, M.C.; Hartlage-Rübsamen, M.; et al. Amyloid-Beta Peptides Trigger Aggregation of Alpha-Synuclein In Vitro. Molecules 2020, 25, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, M.; Miners, J.S.; De Silva, R.; Lashley, T.; Ling, H.; Holton, J.; Revesz, T.; Love, S. Evaluating the relationship between amyloid-β and α-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimers. Res. Ther. 2014, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.H.; Chung, K.C. Death-associated Protein Kinase 1 Phosphorylates α-Synuclein at Ser129 and Exacerbates Rotenone-induced Toxic Aggregation of α-Synuclein in Dopaminergic SH-SY5Y Cells. Exp. Neurobiol. 2020, 29, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Pegueroles, J.; Herrmann, A.G.; Henstridge, C.M.; Muñoz, L.; Querol-Vilaseca, M.; Martín-Paniello, C.S.; Luque-Cabecerans, J.; Clarimon, J.; Belbin, O.; et al. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain 2017, 140, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Deng, M.F.; Xiong, W.; Xie, A.J.; Guo, J.; Liang, Z.H.; Hu, B.; Chen, J.G.; Zhu, X.; Man, H.Y.; et al. MicroRNA-26a/Death-Associated Protein Kinase 1 Signaling Induces Synucleinopathy and Dopaminergic Neuron Degeneration in Parkinson’s Disease. Biol. Psychiatry 2019, 85, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Mahley, R.W. Apolipoprotein E: Cholesterol Transport Protein with Expanding Role in Cell Biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Troutwine, B.R.; Hamid, L.; Lysaker, C.R.; Strope, T.A.; Wilkins, H.M. Apolipoprotein E and Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 496–510. [Google Scholar] [CrossRef]
- Huynh, T.P.V.; Davis, A.A.; Ulrich, J.D.; Holtzman, D.M. Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 2017, 58, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, M.J.; Pankiewicz, J.; Scholtzova, H.; Mehta, P.D.; Prelli, F.; Quartermain, D.; Wisniewski, T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 18787–18792. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Lee, C.Y.D.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ji, Y.; Mehta, P.; Bates, K.A.; Sun, Y.; Wisniewski, T. Blocking the apolipoprotein E/amyloid-β interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J. Alzheimers. Dis. 2011, 24, 269–285. [Google Scholar] [CrossRef]
- Folin, M.; Baiguera, S.; Guidolin, D.; Di Liddo, R.; Grandi, C.; De Carlo, E.; Nussdorfer, G.G.; Parnigotto, P.P. Apolipoprotein-E modulates the cytotoxic effect of beta-amyloid on rat brain endothelium in an isoform-dependent specific manner. Int. J. Mol. Med. 2006, 17, 821–826. [Google Scholar]
- Dafnis, I.; Argyri, L.; Chroni, A. Amyloid-peptide β 42 Enhances the Oligomerization and Neurotoxicity of apoE4: The C-terminal Residues Leu279, Lys282 and Gln284 Modulate the Structural and Functional Properties of apoE4. Neuroscience 2018, 394, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.; Wilcox, J.N.; Pham, K.T.C.; Fremeau, R.T.; Zeviani, M.; Dwork, A.; Soprano, D.R.; Makover, A.; Goodman, D.S.; Zimmerman, E.A. Transthyretin: A choroid plexus-specific transport protein in human brain. The 1986 S. Weir Mitchell award. Neurology 1986, 36, 900–911. [Google Scholar] [CrossRef]
- Saraiva, M.J.M. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum. Mutat. 2001, 17, 493–503. [Google Scholar] [CrossRef]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimers. Dis. 2014, 39, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human serum transthyretin levels correlate inversely with Alzheimer’s disease. J. Alzheimers. Dis. 2011, 25, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Olajide, O.J.; La Rue, C.; Bergdahl, A.; Chapman, C.A. Inhibiting amyloid beta (1–42) peptide-induced mitochondrial dysfunction prevents the degradation of synaptic proteins in the entorhinal cortex. Front. Aging Neurosci. 2022, 14, 960314. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Morsy, A.; Trippier, P.C. Amyloid-Binding Alcohol Dehydrogenase (ABAD) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2019, 62, 4252–4264. [Google Scholar] [CrossRef]
- Borger, E.; Aitken, L.; Muirhead, K.E.A.; Allen, Z.E.; Ainge, J.A.; Conway, S.J.; Gunn-Moore, F.J. Mitochondrial β-amyloid in Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Sirover, M.A. Moonlighting glyceraldehyde-3-phosphate dehydrogenase: Posttranslational modification, protein and nucleic acid interactions in normal cells and in human pathology. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 354–371. [Google Scholar] [CrossRef]
- Naletova, I.; Schmalhausen, E.; Kharitonov, A.; Katrukha, A.; Saso, L.; Caprioli, A.; Muronetz, V. Non-native glyceraldehyde-3-phosphate dehydrogenase can be an intrinsic component of amyloid structures. Biochim. Biophys. Acta-Proteins Proteomics 2008, 1784, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Li, H.; Gao, Z. Amyloid beta modulated the selectivity of heme-catalyzed protein tyrosine nitration: An alternative mechanism for selective protein nitration. J. Biol. Inorg. Chem. 2012, 17, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Földi, I.; Sergeant, N.; Fülöp, L.; Penke, Z.; Janáky, T.; Szücs, M.; Penke, B. Characterization of the interaction between Abeta 1-42 and glyceraldehyde phosphodehydrogenase. J. Pept. Sci. 2008, 14, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.F.; Tsolaki, M.; Mikhaylova, E.R.; Benken, K.A.; Shevtsov, M.A.; Nikotina, A.D.; Lechpammer, M.; Mitkevich, V.A.; Makarov, A.A.; Moskalev, A.A.; et al. Extracellular GAPDH Promotes Alzheimer Disease Progression by Enhancing Amyloid-β Aggregation and Cytotoxicity. Aging Dis. 2021, 12, 1223–1237. [Google Scholar] [CrossRef]
- Orru, S.; Ruoppolo, M.; Francese, S.; Vitagliano, L.; Marino, G.; Esposito, C. Identification of tissue transglutaminase-reactive lysine residues in glyceraldehyde-3-phosphate dehydrogenase. Protein Sci. 2002, 11, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.T.; Lu, J.H. Chaperone-Mediated Autophagy in Neurodegenerative Diseases: Molecular Mechanisms and Pharmacological Opportunities. Cells 2022, 11, 2250. [Google Scholar] [CrossRef]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [Green Version]
- Yakubu, U.M.; Morano, K.A. Suppression of aggregate and amyloid formation by a novel intrinsically disordered region in metazoan Hsp110 chaperones. J. Biol. Chem. 2021, 296, 100567. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, D.H.; Yoon, S.Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Dou, J.; Su, P.; Xu, C.; Wen, Z.; Mao, Z.; Li, W. Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem. Biophys. Res. Commun. 2020, 524, 923. [Google Scholar] [CrossRef]
- Beretta, G.; Shala, A.L. Impact of Heat Shock Proteins in Neurodegeneration: Possible Therapeutical Targets. Ann. Neurosci. 2022, 29, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Sinnige, T.; Yu, A.; Morimoto, R.I. Challenging Proteostasis: Role of the Chaperone Network to Control Aggregation-Prone Proteins in Human Disease. Adv. Exp. Med. Biol. 2020, 1243, 53–68. [Google Scholar] [PubMed]
- Ginsberg, S.D.; Neubert, T.A.; Sharma, S.; Digwal, C.S.; Yan, P.; Timbus, C.; Wang, T.; Chiosis, G. Disease-specific interactome alterations via epichaperomics: The case for Alzheimer’s disease. FEBS J. 2022, 289, 2047–2066. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.C.; Joshi, S.; Wang, T.; Bolaender, A.; Gandu, S.; Koren, J.; Che, A.Y.; Taldone, T.; Yan, P.; Sun, W.; et al. The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun. 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurczak, P.; Groves, P.; Szymanska, A.; Rodziewicz-Motowidlo, S. Human cystatin C monomer, dimer, oligomer, and amyloid structures are related to health and disease. FEBS Lett. 2016, 590, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Jaskolski, M.; Grubb, A. The role of cystatin C in cerebral amyloid angiopathy and stroke: Cell biology and animal models. Brain Pathol. 2006, 16, 60–70. [Google Scholar] [CrossRef]
- Perlenfein, T.J.; Murphy, R.M. Expression, purification, and characterization of human cystatin C monomers and oligomers. Protein Expr. Purif. 2016, 117, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selenica, M.L.; Wang, X.; Ostergaard-Pedersen, L.; Westlind-Danielsson, A.; Grubb, A. Cystatin C reduces the in vitro formation of soluble Abeta1-42 oligomers and protofibrils. Scand. J. Clin. Lab. Investig. 2007, 67, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Pawlik, M.; Sastre, M.; Jung, S.S.; Radvinsky, D.S.; Klein, A.M.; Sommer, J.; Schmidt, S.D.; Nixon, R.A.; Mathews, P.M.; et al. Cystatin C inhibits amyloid-beta deposition in Alzheimer’s disease mouse models. Nat. Genet. 2007, 39, 1440–1442. [Google Scholar] [CrossRef]
- Kaeser, S.A.; Herzig, M.C.; Coomaraswamy, J.; Kilger, E.; Selenica, M.L.; Winkler, D.T.; Staufenbiel, M.; Levy, E.; Grubb, A.; Jucker, M. Cystatin C modulates cerebral beta-amyloidosis. Nat. Genet. 2007, 39, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Tizon, B.; Ribe, E.M.; Mi, W.; Troy, C.M.; Levy, E. Cystatin C protects neuronal cells from amyloid-beta-induced toxicity. J. Alzheimers. Dis. 2010, 19, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Frost, C.V.; Zacharias, M. From monomer to fibril: Abeta-amyloid binding to Aducanumab antibody studied by molecular dynamics simulation. Proteins 2020, 88, 1592–1606. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Rabbani, G.; Choi, I. Use of Peptides for the Management of Alzheimer’s Disease: Diagnosis and Inhibition. Front. Aging Neurosci. 2018, 10, 21. [Google Scholar] [CrossRef]
- Mehrazma, B.; Robinson, M.; Opare, S.K.A.; Petoyan, A.; Lou, J.; Hane, F.T.; Rauk, A.; Leonenko, Z. Pseudo-peptide amyloid-β blocking inhibitors: Molecular dynamics and single molecule force spectroscopy study. Biochim. Biophys. Acta. Proteins Proteom. 2017, 1865, 1707–1718. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, E.H.; Lee, S.C.; Kim, A.R.; Park, H.H.; Son, J.W.; Koh, S.H.; Yoon, M.Y. Development of peptide aptamers as alternatives for antibody in the detection of amyloid-beta 42 aggregates. Anal. Biochem. 2020, 609, 113921. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cho, H.J.; Sen, S.; Arango, A.S.; Huynh, T.T.; Huang, Y.; Bandara, N.; Rogers, B.E.; Tajkhorshid, E.; Mirica, L.M. Amphiphilic Distyrylbenzene Derivatives as Potential Therapeutic and Imaging Agents for Soluble and Insoluble Amyloid β Aggregates in Alzheimer’s Disease. J. Am. Chem. Soc. 2021, 143, 10462–10476. [Google Scholar] [CrossRef]
- Medvedev, A.; Buneeva, O.; Kopylov, A.; Gnedenko, O.; Ivanov, A.; Zgoda, V.; Makarov, A.A. Amyloid-binding proteins: Affinity-based separation, proteomic identification, and optical biosensor validation. Methods Mol. Biol. 2015, 1295, 465–477. [Google Scholar] [PubMed]
- Liu, S.; Park, S.; Allington, G.; Prelli, F.; Sun, Y.; Martá-Ariza, M.; Scholtzova, H.; Biswas, G.; Brown, B.; Verghese, P.B.; et al. Targeting Apolipoprotein E/Amyloid β Binding by Peptoid CPO_Aβ17-21 P Ameliorates Alzheimer’s Disease Related Pathology and Cognitive Decline. Sci. Rep. 2017, 7, 8009. [Google Scholar] [CrossRef]
- Johnson, N.R.; Wang, A.C.J.; Coughlan, C.; Sillau, S.; Lucero, E.; Viltz, L.; Markham, N.; Allen, C.; Dhanasekaran, A.R.; Chial, H.J.; et al. Imipramine and olanzapine block apoE4-catalyzed polymerization of Aβ and show evidence of improving Alzheimer’s disease cognition. Alzheimers. Res. Ther. 2022, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.M.; Kim, B.J.; Shusta, E.V.; Murphy, R.M. Transthyretin Mimetics as Anti-β-Amyloid Agents: A Comparison of Peptide and Protein Approaches. ChemMedChem 2018, 13, 968–979. [Google Scholar] [CrossRef]
- Cao, Q.; Anderson, D.H.; Liang, W.Y.; Chou, J.; Saelices, L. The inhibition of cellular toxicity of amyloid-β by dissociated transthyretin. J. Biol. Chem. 2020, 295, 14015–14024. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Chen, Q.; Zhu, X.; Wang, Y. ABAD/17β-HSD10 reduction contributes to the protective mechanism of huperzine a on the cerebral mitochondrial function in APP/PS1 mice. Neurobiol. Aging 2019, 81, 77–87. [Google Scholar] [CrossRef]
- Morsy, A.; Maddeboina, K.; Gao, J.; Wang, H.; Valdez, J.; Dow, L.F.; Wang, X.; Trippier, P.C. Functionalized Allopurinols Targeting Amyloid-Binding Alcohol Dehydrogenase Rescue Aβ-Induced Mitochondrial Dysfunction. ACS Chem. Neurosci. 2022, 13, 2176–2190. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Benken, K.A.; Semenyuk, P.I.; Sarantseva, S.V.; Bolshakova, O.I.; Mikhaylova, E.R.; Muronetz, V.I.; Guzhova, I.V.; Margulis, B.A. GAPDH binders as potential drugs for the therapy of polyglutamine diseases: Design of a new screening assay. FEBS Lett. 2015, 589, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Lazarev, V.F.; Nikotina, A.D.; Semenyuk, P.I.; Evstafyeva, D.B.; Mikhaylova, E.R.; Muronetz, V.I.; Shevtsov, M.A.; Tolkacheva, A.V.; Dobrodumov, A.V.; Shavarda, A.L.; et al. Small molecules preventing GAPDH aggregation are therapeutically applicable in cell and rat models of oxidative stress. Free Radic. Biol. Med. 2016, 92, 29–38. [Google Scholar] [CrossRef]
- Zatsepina, O.G.; Evgen’ev, M.B.; Garbuz, D.G. Role of a Heat Shock Transcription Factor and the Major Heat Shock Protein Hsp70 in Memory Formation and Neuroprotection. Cells 2021, 10, 1638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, H.; Lobo, N.; Guo, X.; Gentleman, S.M.; Ma, D. Celastrol enhances cell viability and inhibits amyloid-β production induced by lipopolysaccharide in vitro. J. Alzheimers. Dis. 2014, 41, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.R.; Chen, S. Suppression of Alzheimer’s disease-related phenotypes by the heat shock protein 70 inducer, geranylgeranylacetone, in APP/PS1 transgenic mice via the ERK/p38 MAPK signaling pathway. Exp. Ther. Med. 2017, 14, 5267–5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, L.; Calvillo, M.; Luna, F.; Pérez-Severiano, F.; Rubio-Osornio, M.; Guevara, J.; Limón, I.D. 17-AAG improves cognitive process and increases heat shock protein response in a model lesion with Aβ25-35. Neuropeptides 2014, 48, 221–232. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.Q.; Heldt, S.A.; Xu, H.; Liao, F.F. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470. [Google Scholar] [CrossRef] [Green Version]
- Dutysheva, E.A.; Utepova, I.A.; Trestsova, M.A.; Anisimov, A.S.; Charushin, V.N.; Chupakhin, O.N.; Margulis, B.A.; Guzhova, I.V.; Lazarev, V.F. Synthesis and approbation of new neuroprotective chemicals of pyrrolyl- and indolylazine classes in a cell model of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 222, 113577. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Dutysheva, E.A.; Mikhaylova, E.R.; Trestsova, M.A.; Utepova, I.A.; Chupakhin, O.N.; Margulis, B.A.; Guzhova, I.V. Indolylazine Derivative Induces Chaperone Expression in Aged Neural Cells and Prevents the Progression of Alzheimer’s Disease. Molecules 2022, 27, 8950. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Guzhova, I.V.; Margulis, B.A. Glyceraldehyde-3-phosphate dehydrogenase is a multifaceted therapeutic target. Pharmaceutics 2020, 12, 416. [Google Scholar] [CrossRef]


| Protein Name | Sample Source | Potential Function | Reference |
|---|---|---|---|
| Ubiquitin-activating enzyme E1 | Plaques | Ubiquitin-dependent proteosomal degradation of Aβ | [40] |
| GFAP | Plaques | Activation of astrocytic immune response and astrocytic damage | [40] |
| HSP70 | Extracellular vesicles | Involvement in ubiqitin-proteosomal and lysosomal degradation of Aβ, disaggregation of fibrils, and immune response activation | [41] |
| PTGFRN | Extracellular vesicles | Positive regulation of APP procession and Aβ production | [46] |
| GAPDH | Extracellular vesicles | Enhancement of Aβ aggregation and toxicity | [41] |
| TTR | Extracellular vesicles | Blockage of Aβ nucleation | [41] |
| CST3 | Extracellular vesicles | Negative regulation of Aβ oligomers and aggregate formation | [41] |
| Tip60 | Protein–protein interaction | In complex with HDAC2 protection against AD-associated pathologies | [47] |
| Fibulin 1 | Protein–protein interaction | Regulation of APP cleavage | [43] |
| SLC25A4 | Protein–protein interaction | Unknown | [44] |
| EME1 | Protein–protein interaction | Unknown | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarev, V.F.; Dutysheva, E.A.; Kanunikov, I.E.; Guzhova, I.V.; Margulis, B.A. Protein Interactome of Amyloid-β as a Therapeutic Target. Pharmaceuticals 2023, 16, 312. https://doi.org/10.3390/ph16020312
Lazarev VF, Dutysheva EA, Kanunikov IE, Guzhova IV, Margulis BA. Protein Interactome of Amyloid-β as a Therapeutic Target. Pharmaceuticals. 2023; 16(2):312. https://doi.org/10.3390/ph16020312
Chicago/Turabian StyleLazarev, Vladimir F., Elizaveta A. Dutysheva, Igor E. Kanunikov, Irina V. Guzhova, and Boris A. Margulis. 2023. "Protein Interactome of Amyloid-β as a Therapeutic Target" Pharmaceuticals 16, no. 2: 312. https://doi.org/10.3390/ph16020312