
Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy
 , ,
, ,
Abstract
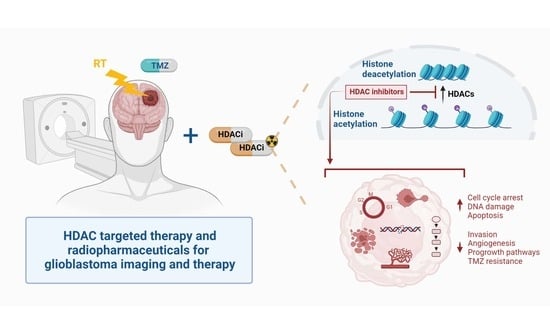
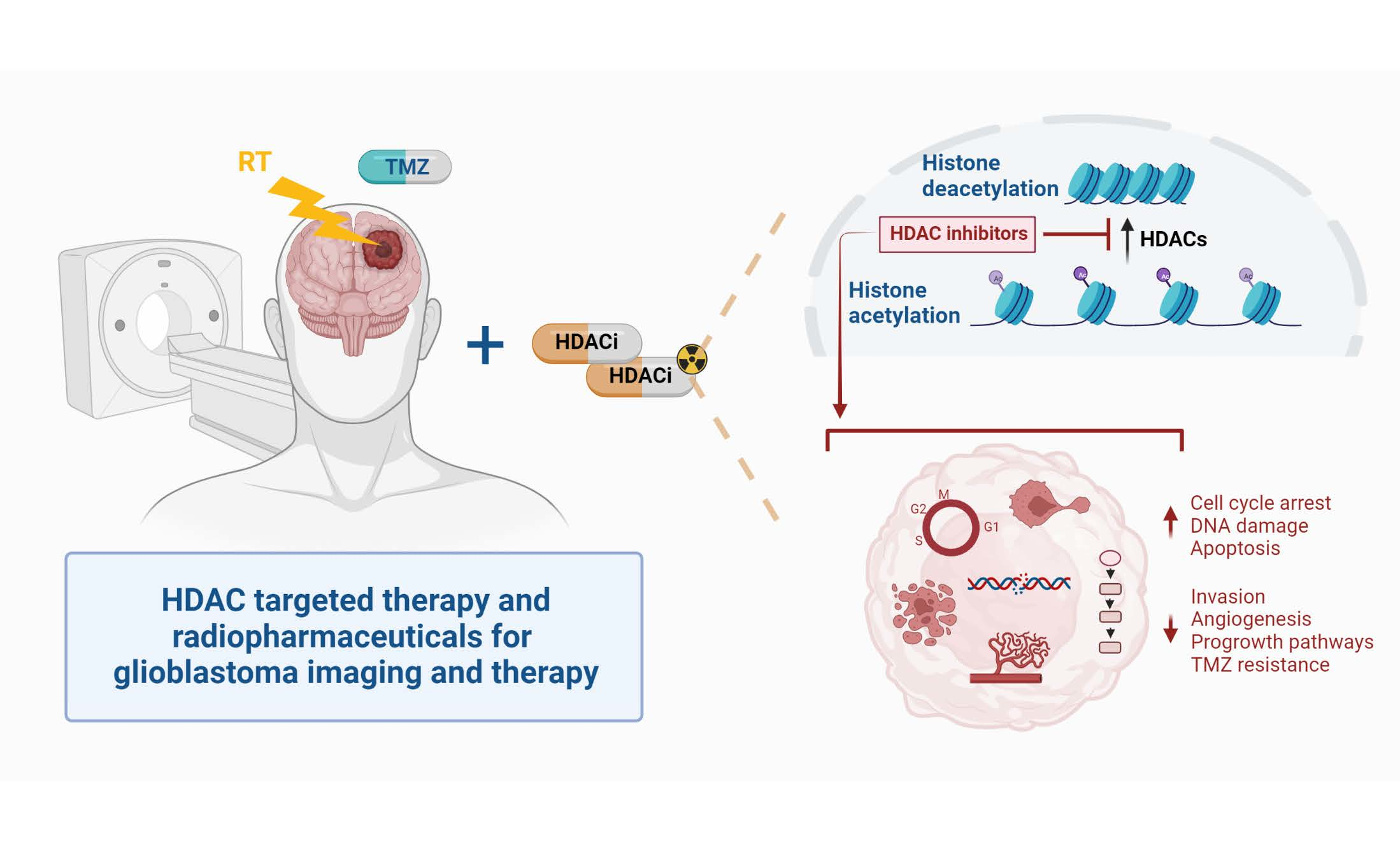
:
1. Introduction
2. Role of HDAC in GB Pathology
3. Current Status of HDACi for GB Therapy
4. HDAC-Targeting Radiopharmaceuticals

{kind=link}
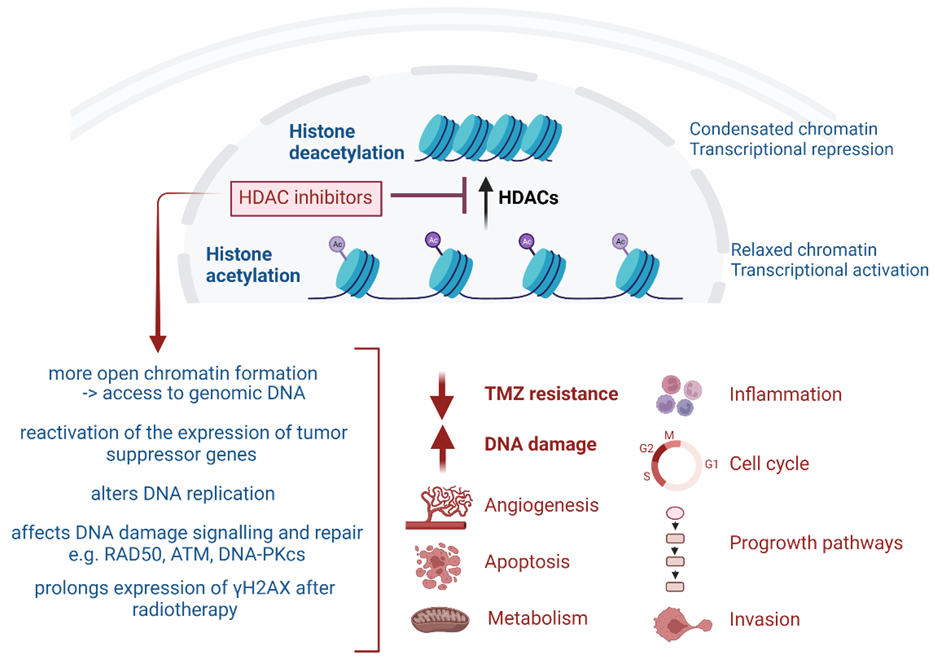{kind=link}
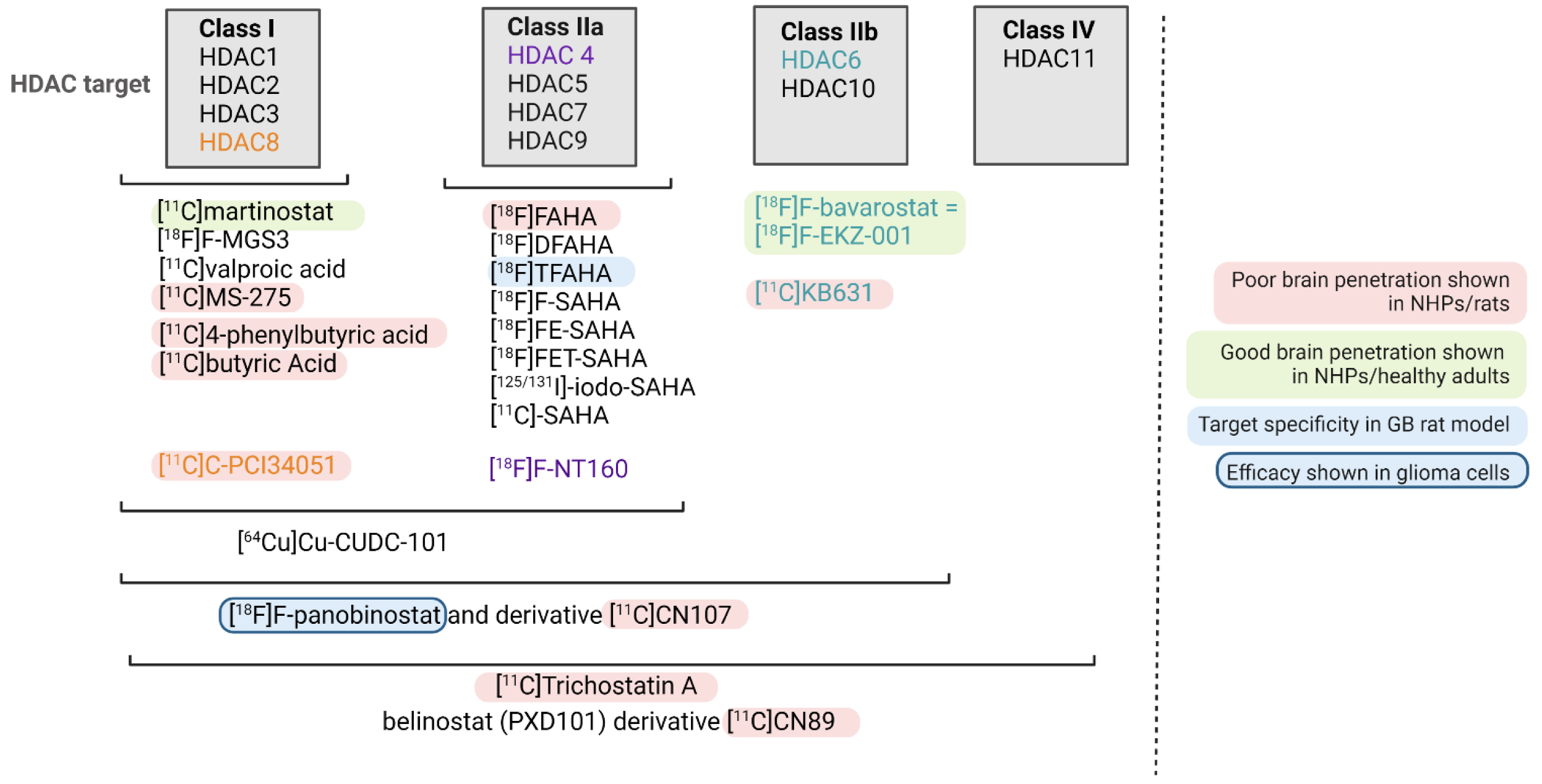{kind=link}
{kind=link}
| Radio- Pharmaceutical | (Pre)clinical Model | Year | Main Outcome and Findings | Ref |
|---|---|---|---|---|
| [18F]FAHA | Healthy rats | 2007 | (+) Uptake increased rapidly up to 0.44%ID/g (5–60 min). Target blocking (SAHA) decreased uptake | [169] |
| Healthy NHP | 2009 | (−) Rapidly metabolized to [18F]FACE, which enters the brain | [171] | |
| Healthy NHP | 2013 | (-) Lack of BBB permeability and specificity | [167] | |
| Healthy NHP | 2013 | (+) BBB crossing. Limited influence of [18F]FACE to brain uptake (first 30 min) | [175] | |
| NNK-treated A/J mice | 2014 | (+) Midbrain, cerebellum and brainstem uptake was displaced by SAHA with <10% remaining | [170] | |
| Healthy mice | 2018 | (+) Specific uptake consistent with increased HDAC levels | [174] | |
| [18F]DFAHA | Healthy rats | 2015 | (+) Selectivity for HDAC Class IIa > [18F]FAHA, favorably low unspecific brain accumulation | [172] |
| [18F]TFAHA | Healthy rats | 2015 | (+) Selectivity for HDAC class IIa > [18F]DFAHA and [18F]FAHA | [172] |
| Intracerebral 9L and U87-MG rat xenografts | 2019 | (+) Increased accumulation at 20 min post-radiotracer administration (+) Target specificity, i.e., significant reduction uptake in 9L tumors after administration of HDACi MC1568 but not the SIRT1 specific inhibitor EX-527 | [163] | |
| AD mouse model | 2021 | (+) Potential as an epigenetic radiotracer for AD | [191] | |
| [18F]F-SAHA | A2780 OC mice | 2011 | (+) Exhibits nM potency. Target binding efficacy can be quantitated within 24 h | [162] |
| [125/131I]-iodo-SAHA | Thyroid, hepatoma, colon carcinoma- bearing mice | 2008 | (−) Equally toxic as SAHA. Rapid efflux and rapid washout and no preferential tumor accumulation. High (unwanted) accumulation in liver and kidneys | [177] |
| [18F]FE-SAHA | Mice LNCaP xenografts | 2011 | (+) Tumor uptake (−) High (unwanted) uptake in small intestines, kidneys, liver and bone (suspected defluorination) | [176] |
| [18F]FET-SAHA | RR1022 sarcoma rat | 2018 | (+) Significant accumulation in tumors with rapid blood clearance (gastrointestinal/renal excretion). Tracer accumulation was receptor-specific | [173] |
| [11C]TSA | Healthy NHP | 2013 | (−) Lack of BBB permeability and HDAC-specificity | [167] |
| [11C]MS-275 | Healthy mice, rats, NHP | 2010 | (−) Poor brain penetration and lack of tracer specificity | [178] |
| [11C]KB631 | B16.F10 murine melanoma-bearing mice | 2019 | (+) Showed HDAC6-selective binding (−) Lack of brain penetrance in rats, possibly due to the hydroxamate moiety | [181] |
| [11C]CN89 | Healthy rats, NHP | 2013 | (−) Poor BBB penetration | [179] |
| [11C]CN107 | ||||
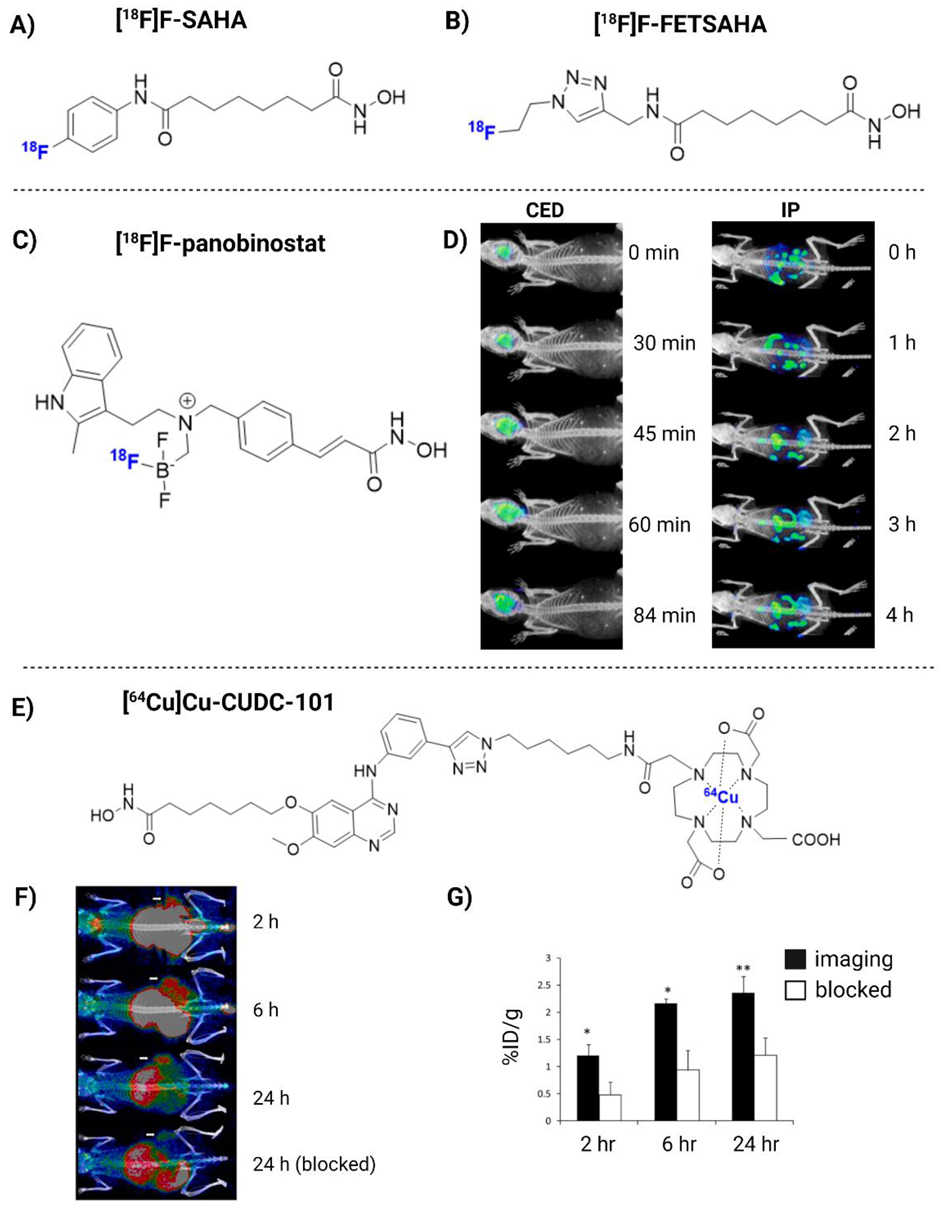
| [18F]F-panobinostat | DIPG IV and XIII + U87MG glioma cells | 2018 | (+) Retains nM efficacy in glioma cells in vitro. Highly selective to glioma, with low toxicity to healthy astrocyte controls. Successful delivery to the murine central nervous system via CED (Figure 3D) | [180] |
| [11C]-4-phenylbutyric acid | Healthy NHP | 2013 | (−) Low brain uptake. Showed 15% metabolization after 30 min. High (unwanted) uptake in liver and heart | [161] |
| [11C]valproic acid | Healthy NHP | 2013 | (−) Low brain uptake. Showed 2% metabolization after 30 min. Exceptionally high (unwanted) heart uptake possibly due to its involvement in lipid metabolism | [161] |
| [11C]butyric Acid | Healthy NHP | 2013 | (−) Low brain uptake. Rapid metabolization (plasma: 80% metabolized after 5 min). Relatively high (unwanted) uptake in spleen and pancreas | [161] |
| [11C]PCI34051 | Healthy rats/NHP | 2013 | (−) Poor BBB penetration. Low uptake in the brain within 80 min. Pretreatment with 2 mg/kg standard did not improve retention or permeability | [179] |
| [64Cu]Cu-CUDC-101 | MDA-MB-231 xenograft mice | 2013 | (+) Specific binding to HDACs in vitro (nM). High TBR in vivo (Figure 3F) | [184] |
| [11C]martinostat | Healthy rats | 2014 | (+) Can quantify target engagement of structurally distinct, brain-penetrant hydroxamate HDACi in living rat brain | [192] |
| Healthy NHP | 2014 | (+) Highly selective and specific. Testing in humans is warranted | [185] | |
| Healthy NHP | 2015 | (+) Allows quantification of brain HDAC expression. Reversible and dose-dependent binding. Slow washout kinetics observed | [193] | |
| Healthy humans | 2016 | (+) Selectively binds HDAC1, 2 and 3 | [186] | |
| Healthy pigs | 2020 | (+) Allowed for accurate in vivo measurement of cerebral HDAC1—3 protein levels. Excellent signal-to-noise ratio | [187] | |
| [18F]F-MGS3 | Healthy rats, NHP | 2016 | (+) Exhibits specific binding/comparable brain uptake and regional distribution to [11C]martinostat | [188] |
| [18F]F-bavarostat | Healthy rats, NHP | 2017 | (+) Selective HDAC6 inhibitor. Excellent brain penetrance. Low amount of nonspecific binding observed after pre-treatment with 1 mg/kg unlabeled bavarostat | [194] |
| Healthy NHP | 2020 | (+) Excellent brain penetrance. Good HDAC6 selectivity, enabling quantification | [189] | |
| Healthy humans | 2021 | (+) Safe to administer and accurate quantification of HDAC6 expression in the human brain | [190] | |
| [18F]F-NT160 | Healthy mice | 2022 | (+) Can cross the BBB and bind to class-IIa HDACs in vivo in mice brain tissue | [183] |
5. Challenges and Future Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. HDAC6 in Diseases of Cognition and of Neurons. Cells 2020, 10, 12. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Pre-clinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Yelton, C.J.; Ray, S.K. Histone deacetylase enzymes and selective histone deacetylase inhibitors for antitumor effects and enhancement of antitumor immunity in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 46. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcène, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, J.M.; Chen, J.K.; Yan, X.L.; Chen, H.M.; Nong, W.X.; Huang, H.Q. Histone deacetylase 5 promotes the proliferation of glioma cells by upregulation of Notch 1. Mol. Med. Rep. 2014, 10, 2045–2050. [Google Scholar] [CrossRef]
- Shi, P.; Hoang-Minh, L.B.; Tian, J.; Cheng, A.; Basrai, R.; Kalaria, N.; Lebowitz, J.J.; Khoshbouei, H.; Deleyrolle, L.P.; Sarkisian, M.R. HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells. Cancers 2021, 13, 1644. [Google Scholar] [CrossRef]
- Li, S.; Liu, X.; Chen, X.; Zhang, L.; Wang, X. Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling. Tumour. Biol. 2015, 36, 9661–9665. [Google Scholar] [CrossRef]
- Huang, Z.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234. [Google Scholar] [CrossRef]
- Kim, G.W.; Lee, D.H.; Yeon, S.K.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Temozolomide-resistant Glioblastoma Depends on HDAC6 Activity Through Regulation of DNA Mismatch Repair. Anticancer Res. 2019, 39, 6731–6741. [Google Scholar] [CrossRef]
- Chueh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 Repression of BCL-XL Determines Apoptotic Sensitivity to HDAC Inhibitors across Tumor Types. Clin. Cancer Res. 2017, 23, 5573–5584. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Lee, I.N.; Huang, C.; Wu, Y.P.; Chung, C.Y.; Lee, M.H.; Lin, M.H.; Yang, J.T. Valproic acid-induced amphiregulin secretion confers resistance to temozolomide treatment in human glioma cells. BMC Cancer 2019, 19, 756. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef]
- Yang, W.B.; Wu, A.C.; Hsu, T.I.; Liou, J.P.; Lo, W.L.; Chang, K.Y.; Chen, P.Y.; Kikkawa, U.; Yang, S.T.; Kao, T.J.; et al. Histone deacetylase 6 acts upstream of DNA damage response activation to support the survival of glioblastoma cells. Cell Death Dis. 2021, 12, 884. [Google Scholar] [CrossRef]
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: Effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef]
- Shabason, J.E.; Tofilon, P.J.; Camphausen, K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J. Cell Mol. Med. 2011, 15, 2735–2744. [Google Scholar] [CrossRef]
- Camphausen, K.; Tofilon, P.J. Inhibition of histone deacetylation: A strategy for tumor radiosensitization. J. Clin. Oncol. 2007, 25, 4051–4056. [Google Scholar] [CrossRef]
- Camphausen, K.; Cerna, D.; Scott, T.; Sproull, M.; Burgan, W.E.; Cerra, M.A.; Fine, H.; Tofilon, P.J. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int. J. Cancer 2005, 114, 380–386. [Google Scholar] [CrossRef]
- Kim, J.H.; Shin, J.H.; Kim, I.H. Susceptibility and radiosensitization of human glioblastoma cells to trichostatin A, a histone deacetylase inhibitor. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 1174–1180. [Google Scholar] [CrossRef]
- Camphausen, K.; Burgan, W.; Cerra, M.; Oswald, K.A.; Trepel, J.B.; Lee, M.J.; Tofilon, P.J. Enhanced radiation-induced cell killing and prolongation of gammaH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res. 2004, 64, 316–321. [Google Scholar] [CrossRef]
- Diss, E.; Nalabothula, N.; Nguyen, D.; Chang, E.; Kwok, Y.; Carrier, F. Vorinostat(SAHA) Promotes Hyper-Radiosensitivity in Wild Type p53 Human Glioblastoma Cells. J. Clin. Oncol. Res. 2014, 2, 1. [Google Scholar]
- Barazzuol, L.; Jeynes, J.C.; Merchant, M.J.; Wéra, A.C.; Barry, M.A.; Kirkby, K.J.; Suzuki, M. Radiosensitization of glioblastoma cells using a histone deacetylase inhibitor (SAHA) comparing carbon ions with X-rays. Int. J. Radiat. Biol. 2015, 91, 90–98. [Google Scholar] [CrossRef]
- Conti, C.; Leo, E.; Eichler, G.S.; Sordet, O.; Martin, M.M.; Fan, A.; Aladjem, M.I.; Pommier, Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010, 70, 4470–4480. [Google Scholar] [CrossRef]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A., 3rd; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef]
- Lee, E.Q.; Puduvalli, V.K.; Reid, J.M.; Kuhn, J.G.; Lamborn, K.R.; Cloughesy, T.F.; Chang, S.M.; Drappatz, J.; Yung, W.K.; Gilbert, M.R.; et al. Phase I study of vorinostat in combination with temozolomide in patients with high-grade gliomas: North American Brain Tumor Consortium Study 04-03. Clin. Cancer Res. 2012, 18, 6032–6039. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 5 September 2022).
- Puduvalli, V.K.; Wu, J.; Yuan, Y.; Armstrong, T.S.; Groves, M.D.; Raizer, J.J.; Giglio, P.; Colman, H.; Peereboom, D.M.; Walbert, T.; et al. Brain Tumor Trials Collaborative Bayesian Adaptive Randomized Phase II trial of bevacizumab plus vorinostat versus bevacizumab alone in adults with recurrent glioblastoma (BTTC-1102). J. Clin. Oncol. 2015, 33, 2012. [Google Scholar] [CrossRef]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neurooncol. 2018, 137, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157-e21. [Google Scholar] [CrossRef]
- Kang, D.W.; Hwang, W.C.; Noh, Y.N.; Kang, Y.; Jang, Y.; Kim, J.A.; Min, D.S. Phospholipase D1 is upregulated by vorinostat and confers resistance to vorinostat in glioblastoma. J. Cell. Physiol. 2021, 236, 549–560. [Google Scholar] [CrossRef]
- Gurbani, S.S.; Yoon, Y.; Weinberg, B.D.; Salgado, E.; Press, R.H.; Cordova, J.S.; Ramesh, K.K.; Liang, Z.; Velazquez Vega, J.; Voloschin, A.; et al. Assessing Treatment Response of Glioblastoma to an HDAC Inhibitor Using Whole-Brain Spectroscopic MRI. Tomography 2019, 5, 53–60. [Google Scholar] [CrossRef]
- Xu, K.; Ramesh, K.; Huang, V.; Gurbani, S.S.; Cordova, J.S.; Schreibmann, E.; Weinberg, B.D.; Sengupta, S.; Voloschin, A.D.; Holdhoff, M.; et al. Final Report on Clinical Outcomes and Tumor Recurrence Patterns of a Pilot Study Assessing Efficacy of Belinostat (PXD-101) with Chemoradiation for Newly Diagnosed Glioblastoma. Tomography 2022, 8, 688–700. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Kuhn, J.G.; Wen, P.Y.; Yung, W.K.; Gilbert, M.R.; Chang, S.M.; Lieberman, F.S.; Prados, M.D.; Fine, H.A. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro-Oncology 2011, 13, 509–516. [Google Scholar] [CrossRef]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Van Veggel, M.; Westerman, E.; Hamberg, P. Clinical Pharmacokinetics and Pharmacodynamics of Panobinostat. Clin. pharmacokinet. 2018, 57, 21–29. [Google Scholar] [CrossRef]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro-Oncology 2015, 17, 862–867. [Google Scholar] [CrossRef]
- Shi, W.; Palmer, J.D.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Bar-Ad, V.; Judy, K.; Farrell, C.; et al. Phase I trial of panobinostat and fractionated stereotactic re-irradiation therapy for recurrent high grade gliomas. J. Neuro-Oncol. 2016, 127, 535–539. [Google Scholar] [CrossRef]
- Singleton, W.G.B.; Bienemann, A.S.; Woolley, M.; Johnson, D.; Lewis, O.; Wyatt, M.J.; Damment, S.J.P.; Boulter, L.J.; Killick-Cole, C.L.; Asby, D.J.; et al. The distribution, clearance, and brainstem toxicity of panobinostat administered by convection-enhanced delivery. J. Neurosurg. Pediatr. 2018, 22, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Guan, W. Valproic Acid: A Promising Therapeutic Agent in Glioma Treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef]
- Tsai, H.C.; Wei, K.C.; Chen, P.Y.; Huang, C.Y.; Chen, K.T.; Lin, Y.J.; Cheng, H.W.; Chen, Y.R.; Wang, H.T. Valproic Acid Enhanced Temozolomide-Induced Anticancer Activity in Human Glioma Through the p53-PUMA Apoptosis Pathway. Front. Oncol. 2021, 11, 722754. [Google Scholar] [CrossRef]
- Krauze, A.V.; Megan, M.; Theresa, C.Z.; Peter, M.; Shih, J.H.; Tofilon, P.J.; Rowe, L.; Gilbert, M.; Camphausen, K. The addition of Valproic acid to concurrent radiation therapy and temozolomide improves patient outcome: A Correlative analysis of RTOG 0525, SEER and a Phase II NCI trial. Cancer Stud. Ther. 2020, 5, 722754. [Google Scholar] [CrossRef]
- Shim, H.; Wei, L.; Holder, C.A.; Guo, Y.; Hu, X.P.; Miller, A.H.; Olson, J.J. Use of high-resolution volumetric MR spectroscopic imaging in assessing treatment response of glioblastoma to an HDAC inhibitor. Am. J. Roentgenol. 2014, 203, W158–W165. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Chowdhary, S.; Potthast, L.; Prabhu, A.; Tsai, Y.Y.; Sarcar, B.; Kahali, S.; Brem, S.; Yu, H.M.; Rojiani, A.; et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro-oncology 2012, 14, 93–100. [Google Scholar] [CrossRef]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro-oncology 2012, 14, 215–221. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-oncology 2018, 20, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kuwabara, Y.; Suehiro, S.; Yamashita, D.; Tanaka, M.; Tanaka, A.; Ohue, S.; Araki, H. Valproic acid reduces hair loss and improves survival in patients receiving temozolomide-based radiation therapy for high-grade glioma. Eur. J. Clin. Pharmacol. 2017, 73, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, H.; Tan, Q.; Xie, C.; Zhan, W.; Sharma, A.; Sharma, H.S.; Zhang, Z. The therapeutic and neuroprotective effects of an antiepileptic drug valproic acid in glioma patients. Prog. Brain Res. 2020, 258, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, M.; Dielemans, J.C.; van Breemen, M.S.; Zwinkels, H.; Walchenbach, R.; Taphoorn, M.J.; Vecht, C.J. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro-oncology 2013, 15, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Valiyaveettil, D.; Malik, M.; Joseph, D.M.; Ahmed, S.F.; Kothwal, S.A.; Vijayasaradhi, M. Effect of valproic acid on survival in glioblastoma: A prospective single-arm study. S. Asian J. Cancer 2018, 7, 159–162. [Google Scholar] [CrossRef]
- Redjal, N.; Reinshagen, C.; Le, A.; Walcott, B.P.; McDonnell, E.; Dietrich, J.; Nahed, B.V. Valproic acid, compared to other antiepileptic drugs, is associated with improved overall and progression-free survival in glioblastoma but worse outcome in grade II/III gliomas treated with temozolomide. J. Neuro-Oncol. 2016, 127, 505–514. [Google Scholar] [CrossRef]
- Ugur, H.C.; Ramakrishna, N.; Bello, L.; Menon, L.G.; Kim, S.K.; Black, P.M.; Carroll, R.S. Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J. Neuro-Oncol. 2007, 83, 267–275. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Addo-Yobo, S.O.; Pollack, I.F. Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J. Pharmacol. Exp. Ther. 2009, 331, 327–337. [Google Scholar] [CrossRef]
- Orzan, F.; Pellegatta, S.; Poliani, P.L.; Pisati, F.; Caldera, V.; Menghi, F.; Kapetis, D.; Marras, C.; Schiffer, D.; Finocchiaro, G. Enhancer of Zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol. Appl. Neurobiol. 2011, 37, 381–394. [Google Scholar] [CrossRef]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro-Oncology 2011, 13, 894–903. [Google Scholar] [CrossRef]
- Berghauser Pont, L.M.; Spoor, J.K.; Venkatesan, S.; Swagemakers, S.; Kloezeman, J.J.; Dirven, C.M.; van der Spek, P.J.; Lamfers, M.L.; Leenstra, S. The Bcl-2 inhibitor Obatoclax overcomes resistance to histone deacetylase inhibitors SAHA and LBH589 as radiosensitizers in patient-derived glioblastoma stem-like cells. Genes Cancer 2014, 5, 445–459. [Google Scholar] [CrossRef]
- Cornago, M.; Garcia-Alberich, C.; Blasco-Angulo, N.; Vall-Llaura, N.; Nager, M.; Herreros, J.; Comella, J.X.; Sanchis, D.; Llovera, M. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014, 5, e1435. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef]
- Lohitesh, K.; Saini, H.; Srivastava, A.; Mukherjee, S.; Roy, A.; Chowdhury, R. Autophagy inhibition potentiates SAHA-mediated apoptosis in glioblastoma cells by accumulation of damaged mitochondria. Oncol. Rep. 2018, 39, 2787–2796. [Google Scholar] [CrossRef]
- Gonçalves, R.M.; Agnes, J.P.; Delgobo, M.; de Souza, P.O.; Thomé, M.P.; Heimfarth, L.; Lenz, G.; Moreira, J.C.F.; Zanotto-Filho, A. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem. Pharmacol. 2019, 163, 440–450. [Google Scholar] [CrossRef]
- Khathayer, F.; Taylor, M.A.; Ray, S.K. Synergism of 4HPR and SAHA increases anti-tumor actions in glioblastoma cells. Apoptosis 2020, 25, 217–232. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, Z.; Copland, J.A.; Mehrling, T.; Tun, H.W. Combined alkylation and histone deacetylase inhibition with EDO-S101 has significant therapeutic activity against brain tumors in preclinical models. Oncotarget 2018, 9, 28155–28164. [Google Scholar] [CrossRef]
- Kusaczuk, M.; Krętowski, R.; Stypułkowska, A.; Cechowska-Pasko, M. Molecular and cellular effects of a novel hydroxamate-based HDAC inhibitor - belinostat - in glioblastoma cell lines: A preliminary report. Investig. New Drugs 2016, 34, 552–564. [Google Scholar] [CrossRef]
- Berghauser Pont, L.M.; Kleijn, A.; Kloezeman, J.J.; van den Bossche, W.; Kaufmann, J.K.; de Vrij, J.; Leenstra, S.; Dirven, C.M.; Lamfers, M.L. The HDAC Inhibitors Scriptaid and LBH589 Combined with the Oncolytic Virus Delta24-RGD Exert Enhanced Anti-Tumor Efficacy in Patient-Derived Glioblastoma Cells. PLoS ONE 2015, 10, e0127058. [Google Scholar] [CrossRef]
- Meng, W.; Wang, B.; Mao, W.; Wang, J.; Zhao, Y.; Li, Q.; Zhang, C.; Tang, Y.; Ma, J. Enhanced efficacy of histone deacetylase inhibitor combined with bromodomain inhibitor in glioblastoma. J. Exp. Clin. Cancer Res. 2018, 37, 241. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, J.; Urdiciain, A.; Zazpe, I.; Zelaya, M.V.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. The synergistic effect of DZ-NEP, panobinostat and temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2020, 56, 283–300. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, J.; Urdiciain, A.; Zelaya, M.V.; Zazpe, I.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. APR-246 combined with 3-deazaneplanocin A, panobinostat or temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2021, 58, 312–330. [Google Scholar] [CrossRef]
- Pratap, U.P.; Sareddy, G.R.; Liu, Z.; Venkata, P.P.; Liu, J.; Tang, W.; Altwegg, K.A.; Ebrahimi, B.; Li, X.; Tekmal, R.R.; et al. Histone deacetylase inhibitors enhance estrogen receptor beta expression and augment agonist-mediated tumor suppression in glioblastoma. Neurooncol. Adv. 2021, 3, vdab099. [Google Scholar] [CrossRef]
- Knüpfer, M.M.; Hernáiz-Driever, P.; Poppenborg, H.; Wolff, J.E.; Cinatl, J. Valproic acid inhibits proliferation and changes expression of CD44 and CD56 of malignant glioma cells in vitro. Anticancer Res. 1998, 18, 3585–3589. [Google Scholar]
- Chavez-Blanco, A.; Perez-Plasencia, C.; Perez-Cardenas, E.; Carrasco-Legleu, C.; Rangel-Lopez, E.; Segura-Pacheco, B.; Taja-Chayeb, L.; Trejo-Becerril, C.; Gonzalez-Fierro, A.; Candelaria, M.; et al. Antineoplastic effects of the DNA methylation inhibitor hydralazine and the histone deacetylase inhibitor valproic acid in cancer cell lines. Cancer Cell Int. 2006, 6, 2. [Google Scholar] [CrossRef]
- Das, C.M.; Aguilera, D.; Vasquez, H.; Prasad, P.; Zhang, M.; Wolff, J.E.; Gopalakrishnan, V. Valproic acid induces p21 and topoisomerase-II (alpha/beta) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J. Neurooncol. 2007, 85, 159–170. [Google Scholar] [CrossRef]
- Papi, A.; Ferreri, A.M.; Rocchi, P.; Guerra, F.; Orlandi, M. Epigenetic modifiers as anticancer drugs: Effectiveness of valproic acid in neural crest-derived tumor cells. Anticancer Res. 2010, 30, 535–540. [Google Scholar]
- Alvarez, A.A.; Field, M.; Bushnev, S.; Longo, M.S.; Sugaya, K. The effects of histone deacetylase inhibitors on glioblastoma-derived stem cells. J. Mol. Neurosci. 2015, 55, 7–20. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Yuan, X.; Hu, Z.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic Acid Promotes Human Glioma U87 Cells Apoptosis and Inhibits Glycogen Synthase Kinase-3β Through ERK/Akt Signaling. Cell Physiol. Biochem. 2016, 39, 2173–2185. [Google Scholar] [CrossRef]
- Chang, Y.L.; Huang, L.C.; Chen, Y.C.; Wang, Y.W.; Hueng, D.Y.; Huang, S.M. The synergistic effects of valproic acid and fluvastatin on apoptosis induction in glioblastoma multiforme cell lines. Int. J. Biochem. Cell Biol. 2017, 92, 155–163. [Google Scholar] [CrossRef]
- Tseng, J.H.; Chen, C.Y.; Chen, P.C.; Hsiao, S.H.; Fan, C.C.; Liang, Y.C.; Chen, C.P. Valproic acid inhibits glioblastoma multiforme cell growth via paraoxonase 2 expression. Oncotarget 2017, 8, 14666–14679. [Google Scholar] [CrossRef]
- Garcia, C.G.; Kahn, S.A.; Geraldo, L.H.M.; Romano, I.; Domith, I.; Silva, D.; Dos Santos Assunção, F.; Ferreira, M.J.; Portugal, C.C.; de Souza, J.M.; et al. Combination Therapy with Sulfasalazine and Valproic Acid Promotes Human Glioblastoma Cell Death Through Imbalance of the Intracellular Oxidative Response. Mol. Neurobiol. 2018, 55, 6816–6833. [Google Scholar] [CrossRef]
- Riva, G.; Cilibrasi, C.; Bazzoni, R.; Cadamuro, M.; Negroni, C.; Butta, V.; Strazzabosco, M.; Dalprà, L.; Lavitrano, M.; Bentivegna, A. Valproic Acid Inhibits Proliferation and Reduces Invasiveness in Glioma Stem Cells Through Wnt/β Catenin Signalling Activation. Genes 2018, 9, 522. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, S.; Frijlink, E.; Kroonen, J.; Spliet, W.G.M.; van Hecke, W.; Seute, T.; Snijders, T.J.; Robe, P.A. Effects of valproic acid on histone deacetylase inhibition in vitro and in glioblastoma patient samples. Neurooncol. Adv. 2019, 1, vdz025. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. The effect of valproic acid on intrinsic, extrinsic, and JAK/STAT pathways in neuroblastoma and glioblastoma cell lines. Res. Pharm. Sci. 2022, 17, 392–409. [Google Scholar] [CrossRef]
- Tarasenko, N.; Chekroun-Setti, H.; Nudelman, A.; Rephaeli, A. Comparison of the anticancer properties of a novel valproic acid prodrug to leading histone deacetylase inhibitors. J. Cell Biochem. 2018, 119, 3417–3428. [Google Scholar] [CrossRef]
- Wetzel, M.; Premkumar, D.R.; Arnold, B.; Pollack, I.F. Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J. Neurosurg. 2005, 103, 549–556. [Google Scholar] [CrossRef]
- Svechnikova, I.; Almqvist, P.M.; Ekström, T.J. HDAC inhibitors effectively induce cell type-specific differentiation in human glioblastoma cell lines of different origin. Int. J. Oncol. 2008, 32, 821–827. [Google Scholar]
- Gao, J.; Chen, T.; Liu, J.; Liu, W.; Hu, G.; Guo, X.; Yin, B.; Gong, Y.; Zhao, J.; Qiang, B.; et al. Loss of NECL1, a novel tumor suppressor, can be restored in glioma by HDAC inhibitor-Trichostatin A through Sp1 binding site. Glia 2009, 57, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Foltz, G.; Yoon, J.G.; Lee, H.; Ma, L.; Tian, Q.; Hood, L.; Madan, A. Epigenetic regulation of wnt pathway antagonists in human glioblastoma multiforme. Genes Cancer 2010, 1, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Höring, E.; Podlech, O.; Silkenstedt, B.; Rota, I.A.; Adamopoulou, E.; Naumann, U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. 2013, 33, 1351–1360. [Google Scholar] [PubMed]
- Sassi Fde, A.; Caesar, L.; Jaeger, M.; Nör, C.; Abujamra, A.L.; Schwartsmann, G.; de Farias, C.B.; Brunetto, A.L.; Lopez, P.L.; Roesler, R. Inhibitory activities of trichostatin a in U87 glioblastoma cells and tumorsphere-derived cells. J. Mol. Neurosci. 2014, 54, 27–40. [Google Scholar] [CrossRef]
- Sun, P.; Xia, S.; Lal, B.; Eberhart, C.G.; Quinones-Hinojosa, A.; Maciaczyk, J.; Matsui, W.; Dimeco, F.; Piccirillo, S.M.; Vescovi, A.L.; et al. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells 2009, 27, 1473–1486. [Google Scholar] [CrossRef]
- Carol, H.; Gorlick, R.; Kolb, E.A.; Morton, C.L.; Manesh, D.M.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wozniak, A.; et al. Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Bouché, M.; Dong, Y.C.; Sheikh, S.; Taing, K.; Saxena, D.; Hsu, J.C.; Chen, M.H.; Salinas, R.D.; Song, H.; Burdick, J.A.; et al. Novel Treatment for Glioblastoma Delivered by a Radiation Responsive and Radiopaque Hydrogel. ACS Biomater. Sci. Eng. 2021, 7, 3209–3220. [Google Scholar] [CrossRef]
- Zhang, W.; Lv, S.; Liu, J.; Zang, Z.; Yin, J.; An, N.; Yang, H.; Song, Y. PCI-24781 down-regulates EZH2 expression and then promotes glioma apoptosis by suppressing the PIK3K/Akt/mTOR pathway. Genet. Mol. Biol. 2014, 37, 716–724. [Google Scholar] [CrossRef]
- Asklund, T.; Appelskog, I.B.; Ammerpohl, O.; Ekström, T.J.; Almqvist, P.M. Histone deacetylase inhibitor 4-phenylbutyrate modulates glial fibrillary acidic protein and connexin 43 expression, and enhances gap-junction communication, in human glioblastoma cells. Eur. J. Cancer 2004, 40, 1073–1081. [Google Scholar] [CrossRef]
- Kusaczuk, M.; Krętowski, R.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyrate-a pan-HDAC inhibitor-suppresses proliferation of glioblastoma LN-229 cell line. Tumour. Biol. 2016, 37, 931–942. [Google Scholar] [CrossRef]
- Engelhard, H.H.; Duncan, H.A.; Kim, S.; Criswell, P.S.; Van Eldik, L. Therapeutic effects of sodium butyrate on glioma cells in vitro and in the rat C6 glioma model. Neurosurgery 2001, 48, 616–624. [Google Scholar] [CrossRef]
- Nakagawa, H.; Sasagawa, S.; Itoh, K. Sodium butyrate induces senescence and inhibits the invasiveness of glioblastoma cells. Oncol. Lett. 2018, 15, 1495–1502. [Google Scholar] [CrossRef]
- Taylor, M.A.; Khathayer, F.; Ray, S.K. Quercetin and Sodium Butyrate Synergistically Increase Apoptosis in Rat C6 and Human T98G Glioblastoma Cells Through Inhibition of Autophagy. Neurochem. Res. 2019, 44, 1715–1725. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Kleszcz, R.; Stasiłowicz-Krzemień, A.; Cielecka-Piontek, J. Sodium Butyrate Enhances Curcuminoids Permeability through the Blood-Brain Barrier, Restores Wnt/β-Catenin Pathway Antagonists Gene Expression and Reduces the Viability of Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 11285. [Google Scholar] [CrossRef]
- Pająk, B.; Siwiak-Niedbalska, E.; Jaśkiewicz, A.; Sołtyka, M.; Zieliński, R.; Domoradzki, T.; Fokt, I.; Skóra, S.; Priebe, W. Synergistic Anticancer Effect of Glycolysis and Histone Deacetylases Inhibitors in a Glioblastoma Model. Biomedicines 2021, 9, 1749. [Google Scholar] [CrossRef]
- Zhang, G.; Gan, Y.H. Synergistic antitumor effects of the combined treatment with an HDAC6 inhibitor and a COX-2 inhibitor through activation of PTEN. Oncol. Rep. 2017, 38, 2657–2666. [Google Scholar] [CrossRef] [Green Version]
- Urdiciain, A.; Erausquin, E.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Tubastatin A, an inhibitor of HDAC6, enhances temozolomide-induced apoptosis and reverses the malignant phenotype of glioblastoma cells. Int. J. Oncol. 2019, 54, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi-Iriarte, J.; Saenz-Antoñanzas, A.; Mikelez-Alonso, I.; Carrasco-Garcia, E.; Tellaetxe-Abete, M.; Lawrie, C.H.; Sampron, N.; Cortajarena, A.L.; Matheu, A. Characterization of a new small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis. 2020, 11, 417. [Google Scholar] [CrossRef]
- Yin, C.; Li, P. Growth Suppression of Glioma Cells Using HDAC6 Inhibitor, Tubacin. Open Med. 2018, 13, 221–226. [Google Scholar] [CrossRef]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target Oncol. 2016, 11, 29–40. [Google Scholar] [CrossRef]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Koul, N.; Joseph, C.; Dixit, D.; Ghosh, S.; Sen, E. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J. Cell. Mol. Med. 2010, 14, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Angeletti, F.; Fossati, G.; Pattarozzi, A.; Würth, R.; Solari, A.; Daga, A.; Masiello, I.; Barbieri, F.; Florio, T.; Comincini, S. Inhibition of the Autophagy Pathway Synergistically Potentiates the Cytotoxic Activity of Givinostat (ITF2357) on Human Glioblastoma Cancer Stem Cells. Front. Mol. Neurosci. 2016, 9, 107. [Google Scholar] [CrossRef]
- Taiarol, L.; Bigogno, C.; Sesana, S.; Kravicz, M.; Viale, F.; Pozzi, E.; Monza, L.; Carozzi, V.A.; Meregalli, C.; Valtorta, S.; et al. Givinostat-Liposomes: Anti-Tumor Effect on 2D and 3D Glioblastoma Models and Pharmacokinetics. Cancers 2022, 14, 2978. [Google Scholar] [CrossRef]
- Marampon, F.; Leoni, F.; Mancini, A.; Pietrantoni, I.; Codenotti, S.; Ferella, L.; Megiorni, F.; Porro, G.; Galbiati, E.; Pozzi, P.; et al. Correction to: Histone deacetylase inhibitor ITF2357 (givinostat) reverts transformed phenotype and counteracts stemness in in vitro and in vivo models of human glioblastoma. J. Cancer Res. Clin. Oncol. 2019, 145, 2411. [Google Scholar] [CrossRef]
- Pont, L.M.; Naipal, K.; Kloezeman, J.J.; Venkatesan, S.; van den Bent, M.; van Gent, D.C.; Dirven, C.M.; Kanaar, R.; Lamfers, M.L.; Leenstra, S. DNA damage response and anti-apoptotic proteins predict radiosensitization efficacy of HDAC inhibitors SAHA and LBH589 in patient-derived glioblastoma cells. Cancer Lett. 2015, 356, 525–535. [Google Scholar] [CrossRef]
- Eyupoglu, I.Y.; Hahnen, E.; Trankle, C.; Savaskan, N.E.; Siebzehnrubl, F.A.; Buslei, R.; Lemke, D.; Wick, W.; Fahlbusch, R.; Blumcke, I. Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS-275. Mol. Cancer Ther. 2006, 5, 1248–1255. [Google Scholar] [CrossRef]
- Buyandelger, B.; Bar, E.E.; Hung, K.S.; Chen, R.M.; Chiang, Y.H.; Liou, J.P.; Huang, H.M.; Wang, J.Y. Histone deacetylase inhibitor MPT0B291 suppresses Glioma Growth in vitro and in vivo partially through acetylation of p53. Int. J. Biol. Sci. 2020, 16, 3184–3199. [Google Scholar] [CrossRef]
- Choi, S.A.; Kwak, P.A.; Park, C.K.; Wang, K.C.; Phi, J.H.; Lee, J.Y.; Lee, C.S.; Lee, J.H.; Kim, S.K. A novel histone deacetylase inhibitor, CKD5, has potent anti-cancer effects in glioblastoma. Oncotarget 2017, 8, 9123–9133. [Google Scholar] [CrossRef]
- Bacon, C.L.; O’Driscoll, E.; Regan, C.M. Valproic acid suppresses G1 phase-dependent sialylation of a 65 kDa glycoprotein in the C6glioma cell cycle. Int. J. Dev. Neurosci. 1998, 15, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sampath, D.; Lang, F.F.; Prabhu, S.; Rao, G.; Fuller, G.N.; Liu, Y.; Puduvalli, V.K. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J. Neurooncol. 2011, 105, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sawa, H.; Murakami, H.; Kumagai, M.; Nakasato, M.; Yamauchi, S.; Matsuyama, N.; Tamura, Y.; Satone, A.; Ide, W.; Hashimoto, I.; et al. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004, 107, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Franco-Molina, M.A.; Santana-Krímskaya, S.E.; Madrigal-de-León, L.M.; Coronado-Cerda, E.E.; Zárate-Triviño, D.G.; Hernández-Martínez, S.P.; García-Coronado, P.L.; Rodríguez-Padilla, C. Evaluation of the cytotoxic and immunogenic potential of temozolamide, panobinostat, and Lophophora williamsii extract against C6 glioma cells. Excli. J. 2021, 20, 614–624. [Google Scholar] [CrossRef]
- Hsu, Y.F.; Sheu, J.R.; Hsiao, G.; Lin, C.H.; Chang, T.H.; Chiu, P.T.; Wang, C.Y.; Hsu, M.J. p53 in trichostatin A induced C6 glioma cell death. Biochim. Biophys. Acta 2011, 1810, 504–513. [Google Scholar] [CrossRef]
- Staberg, M.; Michaelsen, S.R.; Rasmussen, R.D.; Villingshoj, M.; Poulsen, H.S.; Hamerlik, P. Inhibition of histone deacetylases sensitizes glioblastoma cells to lomustine. Cell. Oncol. 2017, 40, 21–32. [Google Scholar] [CrossRef]
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140. [Google Scholar] [CrossRef]
- Yu, C.; Friday, B.B.; Yang, L.; Atadja, P.; Wigle, D.; Sarkaria, J.; Adjei, A.A. Mitochondrial Bax translocation partially mediates synergistic cytotoxicity between histone deacetylase inhibitors and proteasome inhibitors in glioma cells. Neuro-Oncology 2008, 10, 309–319. [Google Scholar] [CrossRef]
- Bangert, A.; Hacker, S.; Cristofanon, S.; Debatin, K.M.; Fulda, S. Chemosensitization of glioblastoma cells by the histone deacetylase inhibitor MS275. Anticancer Drugs 2011, 22, 494–499. [Google Scholar] [CrossRef]
- Vengoji, R.; Atri, P.; Macha, M.A.; Seshacharyulu, P.; Perumal, N.; Mallya, K.; Liu, Y.; Smith, L.M.; Rachagani, S.; Mahapatra, S.; et al. Differential gene expression-based connectivity mapping identified novel drug candidate and improved Temozolomide efficacy for Glioblastoma. J. Exp. Clin. Cancer Res. 2021, 40, 335. [Google Scholar] [CrossRef]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Wang, B.; Zhang, X.J.; Li, W.P. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-κB-Dependent Transcription in Glioblastoma Cell Lines. Neurochem. Res. 2016, 41, 3192–3205. [Google Scholar] [CrossRef]
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajic, Z.; Petrecca, K.; Maysinger, D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov. 2018, 4, 41. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef]
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef]
- Yao, Z.G.; Li, W.H.; Hua, F.; Cheng, H.X.; Zhao, M.Q.; Sun, X.C.; Qin, Y.J.; Li, J.M. LBH589 Inhibits Glioblastoma Growth and Angiogenesis Through Suppression of HIF-1α Expression. J. Neuropathol. Exp. Neurol. 2017, 76, 1000–1007. [Google Scholar] [CrossRef]
- An, Z.; Gluck, C.B.; Choy, M.L.; Kaufman, L.J. Suberoylanilide hydroxamic acid limits migration and invasion of glioma cells in two and three dimensional culture. Cancer Lett. 2010, 292, 215–227. [Google Scholar] [CrossRef]
- Perez, T.; Bergès, R.; Maccario, H.; Oddoux, S.; Honoré, S. Low concentrations of vorinostat decrease EB1 expression in GBM cells and affect microtubule dynamics, cell survival and migration. Oncotarget 2021, 12, 304–315. [Google Scholar] [CrossRef]
- Rampazzo, E.; Manfreda, L.; Bresolin, S.; Cani, A.; Mariotto, E.; Bortolozzi, R.; Della Puppa, A.; Viola, G.; Persano, L. Histone Deacetylase Inhibitors Impair Glioblastoma Cell Motility and Proliferation. Cancers 2022, 14, 1897. [Google Scholar] [CrossRef]
- Eyupoglu, I.Y.; Hahnen, E.; Buslei, R.; Siebzehnrubl, F.A.; Savaskan, N.E.; Luders, M.; Trankle, C.; Wick, W.; Weller, M.; Fahlbusch, R.; et al. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J. Neurochem. 2005, 93, 992–999. [Google Scholar] [CrossRef]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef]
- Alexanian, A.R.; Brannon, A. Unique combinations of epigenetic modifiers synergistically impair the viability of the U87 glioblastoma cell line while exhibiting minor or moderate effects on normal stem cell growth. Med. Oncol. 2022, 39, 86. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Shang, E.; Schiffgens, S.; Torrini, C.; Shu, C.; Akman, H.O.; Prabhu, V.V.; Allen, J.E.; Westhoff, M.A.; Karpel-Massler, G.; et al. Induction of Synthetic Lethality by Activation of Mitochondrial ClpP and Inhibition of HDAC1/2 in Glioblastoma. Clin. Cancer Res. 2022, 28, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.; Jane, E.P.; Agostino, N.R.; DiDomenico, J.D.; Pollack, I.F. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol. Carcinog. 2013, 52, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Wang, B.; Mao, W.; Wang, J.; Zhao, Y.; Li, Q.; Zhang, C.; Ma, J. Enhanced efficacy of histone deacetylase inhibitor panobinostat combined with dual PI3K/mTOR inhibitor BEZ235 against glioblastoma. Nagoya J. Med. Sci. 2019, 81, 93–102. [Google Scholar] [CrossRef]
- Essien, E.I.; Hofer, T.P.; Atkinson, M.J.; Anastasov, N. Combining HDAC and MEK Inhibitors with Radiation against Glioblastoma-Derived Spheres. Cells 2022, 11, 775. [Google Scholar] [CrossRef]
- Marino, A.M.; Sofiadis, A.; Baryawno, N.; Johnsen, J.I.; Larsson, C.; Vukojevic, V.; Ekstrom, T.J. Enhanced effects by 4-phenylbutyrate in combination with RTK inhibitors on proliferation in brain tumor cell models. Biochem. Biophys. Res. Commun. 2011, 411, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ishida, C.T.; Ishida, W.; Lo, S.L.; Zhao, J.; Shu, C.; Bianchetti, E.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; et al. Combined HDAC and Bromodomain Protein Inhibition Reprograms Tumor Cell Metabolism and Elicits Synthetic Lethality in Glioblastoma. Clin. Cancer Res. 2018, 24, 3941–3954. [Google Scholar] [CrossRef]
- Kim, G.H.; Choi, S.Y.; Oh, T.I.; Kan, S.Y.; Kang, H.; Lee, S.; Oh, T.; Ko, H.M.; Lim, J.H. IDH1(R132H) Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 2679. [Google Scholar] [CrossRef]
- Wang, C.; Schroeder, F.A.; Hooker, J.M. Visualizing epigenetics: Current advances and advantages in HDAC PET imaging techniques. Neuroscience 2014, 264, 186–197. [Google Scholar] [CrossRef]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. J. Nucl. Med. 2013, 40, 912–918. [Google Scholar] [CrossRef]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef]
- Laws, M.T.; Bonomi, R.E.; Kamal, S.; Gelovani, D.J.; Llaniguez, J.; Potukutchi, S.; Lu, X.; Mangner, T.; Gelovani, J.G. Molecular imaging HDACs class IIa expression-activity and pharmacologic inhibition in intracerebral glioma models in rats using PET/CT/(MRI) with [(18)F]TFAHA. Sci. Rep. 2019, 9, 3595. [Google Scholar] [CrossRef]
- Tago, T.; Toyohara, J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules 2018, 23, 300. [Google Scholar] [CrossRef]
- El Bahhaj, F.; Denis, I.; Pichavant, L.; Delatouche, R.; Collette, F.; Linot, C.; Pouliquen, D.; Grégoire, M.; Héroguez, V.; Blanquart, C.; et al. Histone Deacetylase Inhibitors Delivery using Nanoparticles with Intrinsic Passive Tumor Targeting Properties for Tumor Therapy. Theranostics 2016, 6, 795–807. [Google Scholar] [CrossRef]
- Bolcaen, J.; Kleynhans, J.; Nair, S.; Verhoeven, J.; Goethals, I.; Sathekge, M.; Vandevoorde, C.; Ebenhan, T. A perspective on the radiopharmaceutical requirements for imaging and therapy of glioblastoma. Theranostics 2021, 11, 7911–7947. [Google Scholar] [CrossRef]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorg. Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, U.; Tong, W.P.; Gelovani, J.G.; Alauddin, M.M. Radiosynthesis of 6-([18F]fluoroacetamido)-1-hexanoicanilide ([18F]FAHA) for PET imaging of histone deacetylase (HDAC). J. Label Compd. Radiopharm. 2006, 49, 997–1006. [Google Scholar] [CrossRef]
- Nishii, R.; Mukhopadhyay, U.; Yeh, H.; Soghomonyan, S.; Volgin, A.; Alauddin, M.; Tong, W.; Gelovani, J. PET imaging of histone deacetylase activity in a rat brain using 6-([18F]-fluoroacetamide)-1-hexanoicanilide ([18F]-FAHA). J. Nucl. Med. 2007, 48, 336P. [Google Scholar]
- Tang, W.; Kuruvilla, S.A.; Galitovskiy, V.; Pan, M.L.; Grando, S.A.; Mukherjee, J. Targeting histone deacetylase in lung cancer for early diagnosis: (18)F-FAHA PET/CT imaging of NNK-treated A/J mice model. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 324–332. [Google Scholar]
- Reid, A.E.; Hooker, J.; Shumay, E.; Logan, J.; Shea, C.; Kim, S.W.; Collins, S.; Xu, Y.; Volkow, N.; Fowler, J.S. Evaluation of 6-([(18)F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. J. Nucl. Med. 2009, 36, 247–258. [Google Scholar] [CrossRef]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Kim, H.S.; Kim, M.; Kwon, J.; Kim, E.M.; Hwang, H.; Oh, P.S.; Lim, S.T.; Sohn, M.H.; Kim, D.H.; et al. Synthesis and Evaluation of 2-[(18)F]Fluoroethyltriazolesuberohydroxamine Acid for Histone Deacetylase in a Tumor Model as a Positron Emission Tomography Radiotracer. Cancer Biother Radiopharm 2018, 33, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, N.; Yeh, S.H.; Flores Ii, L.G.; Mukhopadhyay, U.; Young, D.; Ogawa, K.; Jeong, H.J.; Tong, W.; Gelovani, J.G. In Vivo 6-([(18)F]Fluoroacetamido)-1-hexanoicanilide PET Imaging of Altered Histone Deacetylase Activity in Chemotherapy-Induced Neurotoxicity. Contrast Media Mol. Imaging 2018, 2018, 3612027. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Tian, M.; Hinz, R.; Young, D.; Shavrin, A.; Mukhapadhyay, U.; Flores, L.G.; Balatoni, J.; Soghomonyan, S.; Jeong, H.J.; et al. Imaging epigenetic regulation by histone deacetylases in the brain using PET/MRI with (1)(8)F-FAHA. Neuroimage 2013, 64, 630–639. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Pillarsetty, N.; Divilov, V.; Blasberg, R.A.; Lewis, J.S. The synthesis and evaluation of N1-(4-(2-[18F]-fluoroethyl)phenyl)-N8-hydroxyoctanediamide ([18F]-FESAHA), a PET radiotracer designed for the delineation of histone deacetylase expression in cancer. J. Nucl. Med. 2011, 38, 683–696. [Google Scholar] [CrossRef]
- Haberkorn, U.; Beijer, B.; Altmann, A.; Gelovani, J.; Strauss, L.; Dimitrakopoulou-Strauss, A.; Eisenhut, M.; Mier, W. Uptake and biodistribution of the histone deacetylase inhibitor SAHA in tumor bearing animals. J. Nucl. Med. 2008, 49, 332P. [Google Scholar]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef]
- Wang, W.J.; Long, L.M.; Yang, N.; Zhang, Q.Q.; Ji, W.J.; Zhao, J.H.; Qin, Z.H.; Wang, Z.; Chen, G.; Liang, Z.Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef]
- Kommidi, H.; Tosi, U.; Maachani, U.B.; Guo, H.; Marnell, C.S.; Law, B.; Souweidane, M.M.; Ting, R. (18)F-Radiolabeled Panobinostat Allows for Positron Emission Tomography Guided Delivery of a Histone Deacetylase Inhibitor. ACS Med. Chem. Lett. 2018, 9, 114–119. [Google Scholar] [CrossRef]
- Vermeulen, K.; Ahamed, M.; Luyten, K.; Bormans, G. Evaluation of [(11)C]KB631 as a PET tracer for in vivo visualisation of HDAC6 in B16.F10 melanoma. J. Nucl. Med. 2019, 74-75, 1–11. [Google Scholar] [CrossRef]
- Tosi, U.; Kommidi, H.; Adeuyan, O.; Guo, H.; Maachani, U.B.; Chen, N.; Su, T.; Zhang, G.; Pisapia, D.J.; Dahmane, N.; et al. PET, image-guided HDAC inhibition of pediatric diffuse midline glioma improves survival in murine models. Sci. Adv. 2020, 6, eabb4105. [Google Scholar] [CrossRef]
- Turkman, N.; Liu, D.; Pirola, I. Design, synthesis, biochemical evaluation, radiolabeling and in vivo imaging with high affinity class-IIa histone deacetylase inhibitor for molecular imaging and targeted therapy. Eur. J. Med. Chem. 2022, 228, 114011. [Google Scholar] [CrossRef]
- Meng, Q.; Li, F.; Jiang, S.; Li, Z. Novel (64)Cu-Labeled CUDC-101 for in Vivo PET Imaging of Histone Deacetylases. ACS Med. Chem. Lett. 2013, 4, 858–862. [Google Scholar] [CrossRef]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef]
- Donovan, L.L.; Magnussen, J.H.; Dyssegaard, A.; Lehel, S.; Hooker, J.M.; Knudsen, G.M.; Hansen, H.D. Imaging HDACs In Vivo: Cross-Validation of the [(11)C]Martinostat Radioligand in the Pig Brain. Mol. Imaging Biol. 2020, 22, 569–577. [Google Scholar] [CrossRef]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef]
- Fang, X.T.; Zheng, M.Q.; Holden, D.; Fowles, K.; Tamagnan, G.; Hooker, J.; Huang, Y.Y.; Carson, R. Assessment of HDAC6 PET radiotracer F-18-Bavarostat. J. Nucl. Med. 2020, 61, 1021. [Google Scholar]
- Koole, M.; Van Weehaeghe, D.; Serdons, K.; Herbots, M.; Cawthorne, C.; Celen, S.; Schroeder, F.A.; Hooker, J.M.; Bormans, G.; de Hoon, J.; et al. Clinical validation of the novel HDAC6 radiotracer [(18)F]EKZ-001 in the human brain. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 596–611. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lu, C.H.; Ke, C.C.; Chiu, S.J.; Chang, C.W.; Yang, B.H.; Gelovani, J.G.; Liu, R.S. Evaluation of Class IIa Histone Deacetylases Expression and In Vivo Epigenetic Imaging in a Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8633. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Wang, C.; Van de Bittner, G.C.; Neelamegam, R.; Takakura, W.R.; Karunakaran, A.; Wey, H.Y.; Reis, S.A.; Gale, J.; Zhang, Y.L.; et al. PET imaging demonstrates histone deacetylase target engagement and clarifies brain penetrance of known and novel small molecule inhibitors in rat. ACS Chem. Neurosci. 2014, 5, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Wey, H.Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [(1)(1)C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [(18)F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Regimen | Stage | GB Type | Main Result | Reference | ||
|---|---|---|---|---|---|---|
| Vorinostat (SAHA, Zolinza, MK0683) Pan-HDACi | (+) | / | C(II) | Well-tolerated. Modest single-agent activity. Trials with combination regimens warranted | [40] | |
| TMZ | NA | rec/prog | MRS imaging may enable quantitative analysis of tumor response | [59] NCT01342757 * | ||
| TMZ | C(I) | HGG | Well-tolerated | [41] | ||
| BEV/Irinotecan | C(I) | rec | (+) Well-tolerated (+) OS and PFS at 400 mg daily or 300 mg twice a day | NCT00762255 * | ||
| (−) | BEV | C(II) | rec | PFS6 or median OS was not improved | [45] NCT01738646 * | |
| BEV | C(I/II) | rec | Did not improve PFS or OS | [43] NCT01266031 * | ||
| BEV/TMZ | C(I/II) | PFS6 was not statistically improved beyond controls | [44] NCT00939991 * | |||
| BEV/CPT-11 | C(I) | rec | Increased toxicities | [60] | ||
| Erlotinib/BEV | C(I/II) | rec | Trial terminated (toxicities) | NCT01110876 * | ||
| Bortezomib | C(II) | rec | Trial closed at interim analysis (0/34 progression-free) | [61] NCT00641706 * | ||
| FSRT | C(I) | rec | Trial terminated | NCT01378481 * | ||
| TMZ/RT | C(I/II) | nd | Acceptable tolerability, but primary efficacy endpoint not met. Sensitivity signatures could facilitate patient selection | [62] NCT00731731 * | ||
| Ongoing | TMZ | C(I) | HGG | Active: not recruiting | NCT00268385 * | |
| TMZ/Carboplatin/Isotretinoin | C(I/II) | rec | Active: not recruiting | NCT00555399 * | ||
| Pembrolizumab/TMZ/RT | C(I) | nd | Active: recruiting | NCT03426891 * | ||
| Belinostat (PXD101, Beleodaq) Pan-HDACi | Ongoing | TMZ/RT | Pilot study | nd | Active: not recruiting Radiosensitizing effect | [47,48] NCT02137759 * |
| Romidepsin (Istodax, FK228, FR901228, depsipeptide) HDAC class I | (−) | C(I/II) | rec | Ineffective | [49] NCT00085540 * | |
| Ongoing | C(I) | glioma | Active: not recruiting | NCT01638533 * | ||
| Panobinostat (LBH589) HDAC class I/II | (+) | FSRT | C(I) | recHGG | Well-tolerated. Phase II trial warranted | [53] |
| (−) | BEV | C(II) | recHGG | Did not improve PFS6 compared to BEV monotherapy | [52] NCT00859222 * | |
| C(II) | recHGG | Trial terminated due to insufficient accrual | NCT00848523 * | |||
| C(II) | rec | Trial withdrawn due to no enrollment | NCT01115036 * | |||
| Valproic acid (VPA, valproate, Depakene) HDAC class I | (+) | C(II) | HGG | Well-tolerated. May result in improved outcomes. A phase III should follow | [56] NCT00302159 * | |
| HGG | Delayed hair loss and improvement in survival | [63] | ||||
| HGG | Improvement in survival | [64] | ||||
| nd | Improvement in PFS and OS confirmed | [58] | ||||
| nd | Survival benefit dependent on their p53 gene status | [57] | ||||
| Levetiracetam/TMZ/RT | Retro | GB | 2-months longer survival | [65] | ||
| (−) | Doxorubicin/TMZ/RT | C(II) | nd | Trials terminated | NCT02758366 * | |
| Celecoxib | C(II) | Nd | NCT00068770 * | |||
| SRS/Nivolumab | C(I) | Rec | NCT02648633 * | |||
| psa | GB | PFS and OS were comparable to historical controls | [66] | |||
| TMZ | Retro | II/III | VPA was linked to histological progression and decrease in PFS | [67] | ||
| Ongoing | Sildenafil/Sorafenib | C(II) | recHGG | Active: not recruiting | NCT01817751 * | |
| Levetiracetam | C(IV) | glioma | Recruiting: for seizure treatment | NCT03048084 * | ||
| Perampanel | C(IV) | HGG | Recruiting: for seizure treatment | NCT04650204 * | ||
| TMZ | C(III) | HGG | Recruiting | NCT03243461 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Everix, L.; Seane, E.N.; Ebenhan, T.; Goethals, I.; Bolcaen, J. Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy. Pharmaceuticals 2023, 16, 227. https://doi.org/10.3390/ph16020227
Everix L, Seane EN, Ebenhan T, Goethals I, Bolcaen J. Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy. Pharmaceuticals. 2023; 16(2):227. https://doi.org/10.3390/ph16020227
Chicago/Turabian StyleEverix, Liesbeth, Elsie Neo Seane, Thomas Ebenhan, Ingeborg Goethals, and Julie Bolcaen. 2023. "Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy" Pharmaceuticals 16, no. 2: 227. https://doi.org/10.3390/ph16020227