
Properly Substituted Benzimidazoles as a New Promising Class of Nicotinate Phosphoribosyltransferase (NAPRT) Modulators
 , ,
, ,  , , ,
, , ,  ,
,  and
and
Abstract
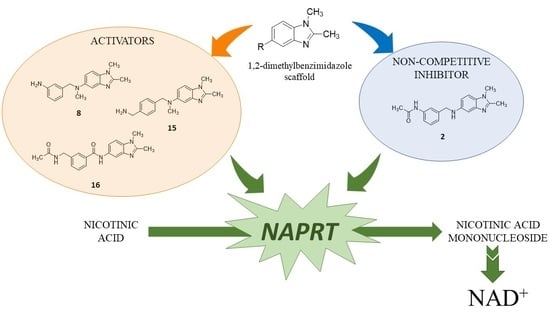
:
1. Introduction
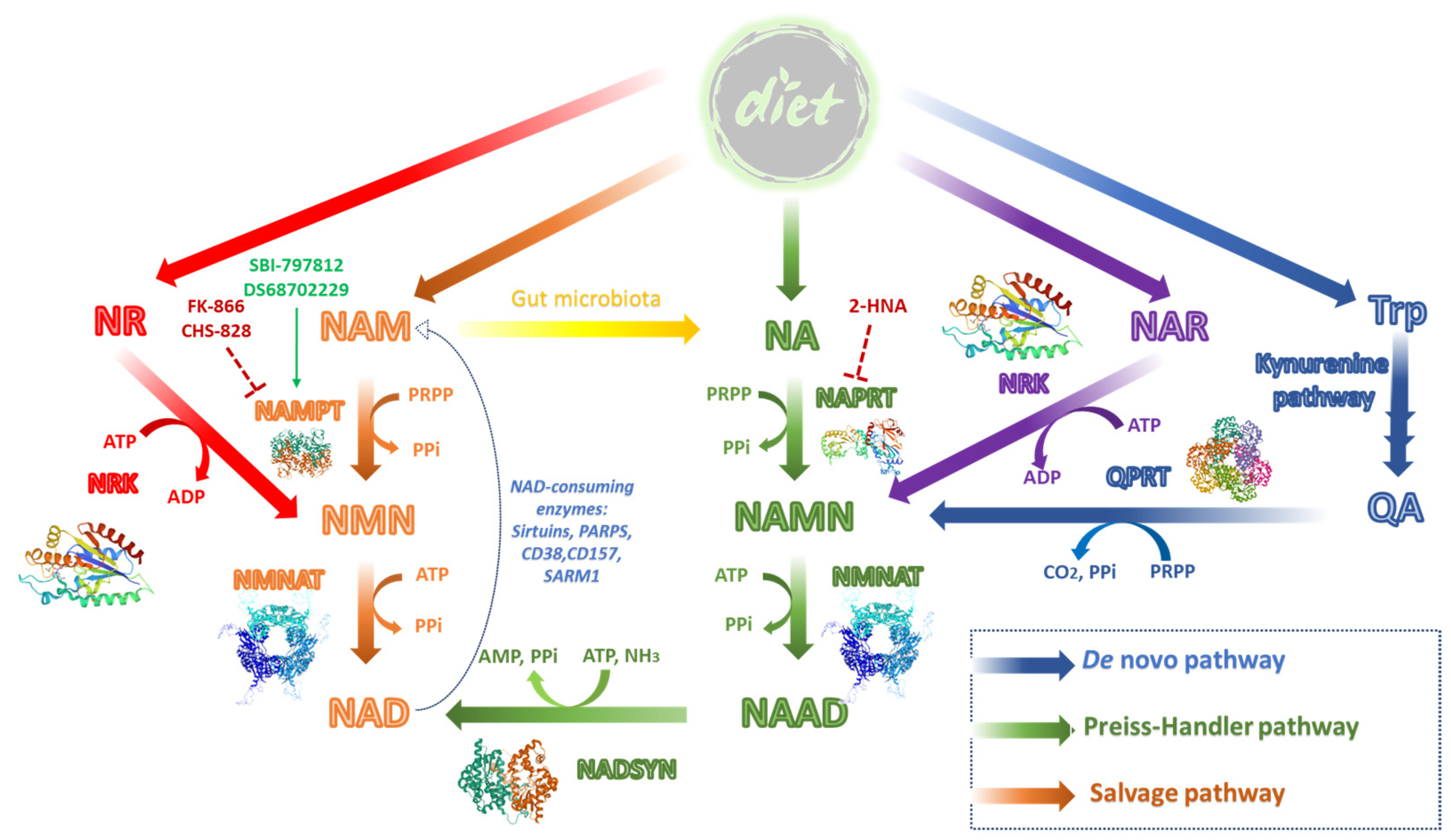
2. Results and Discussion
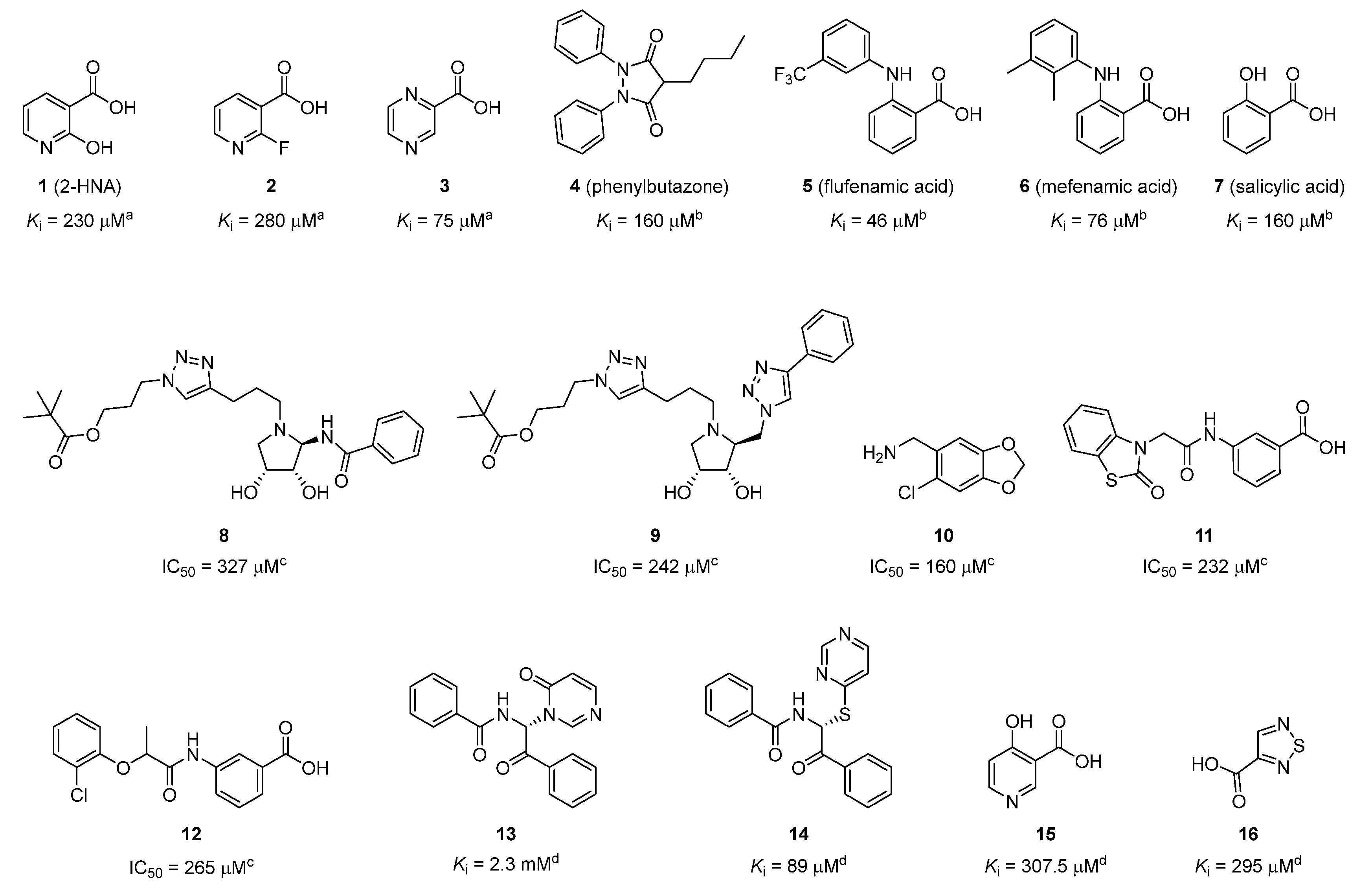
2.1. Identification of the Hit Compound
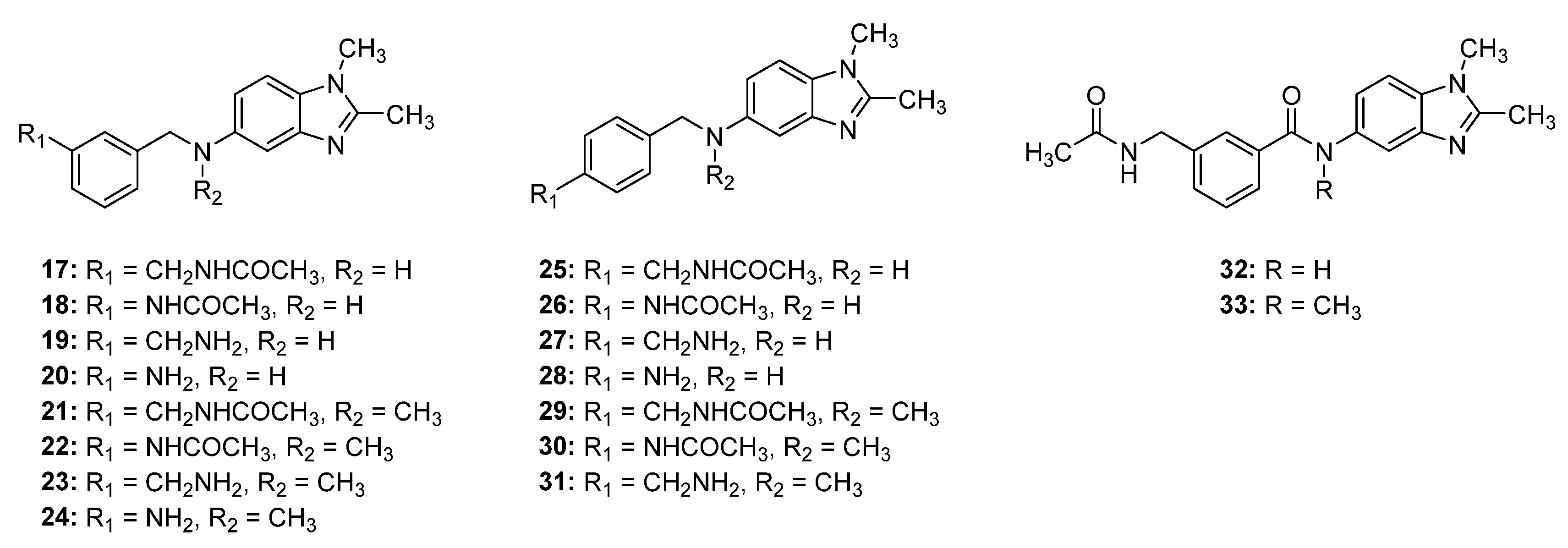
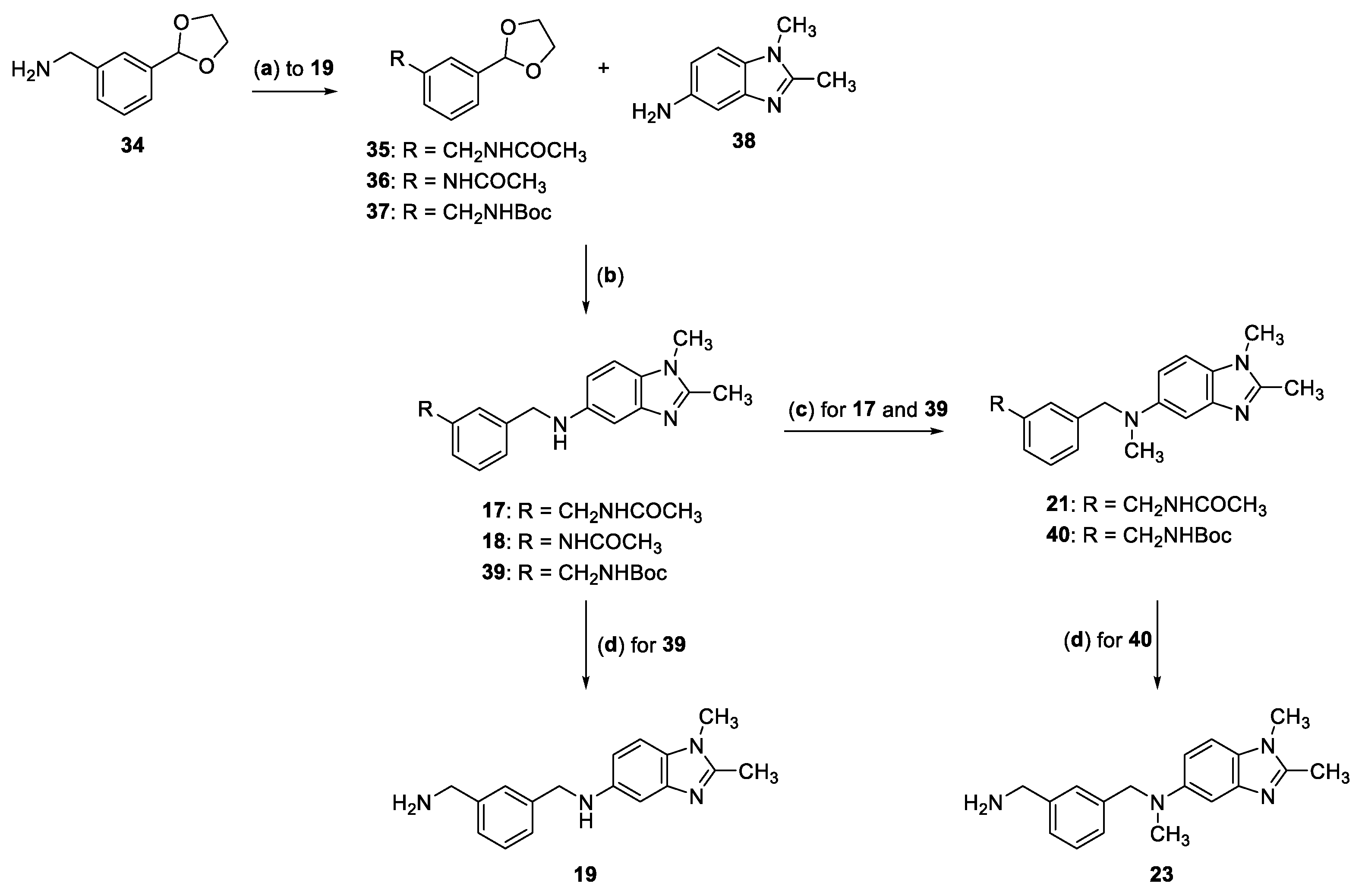
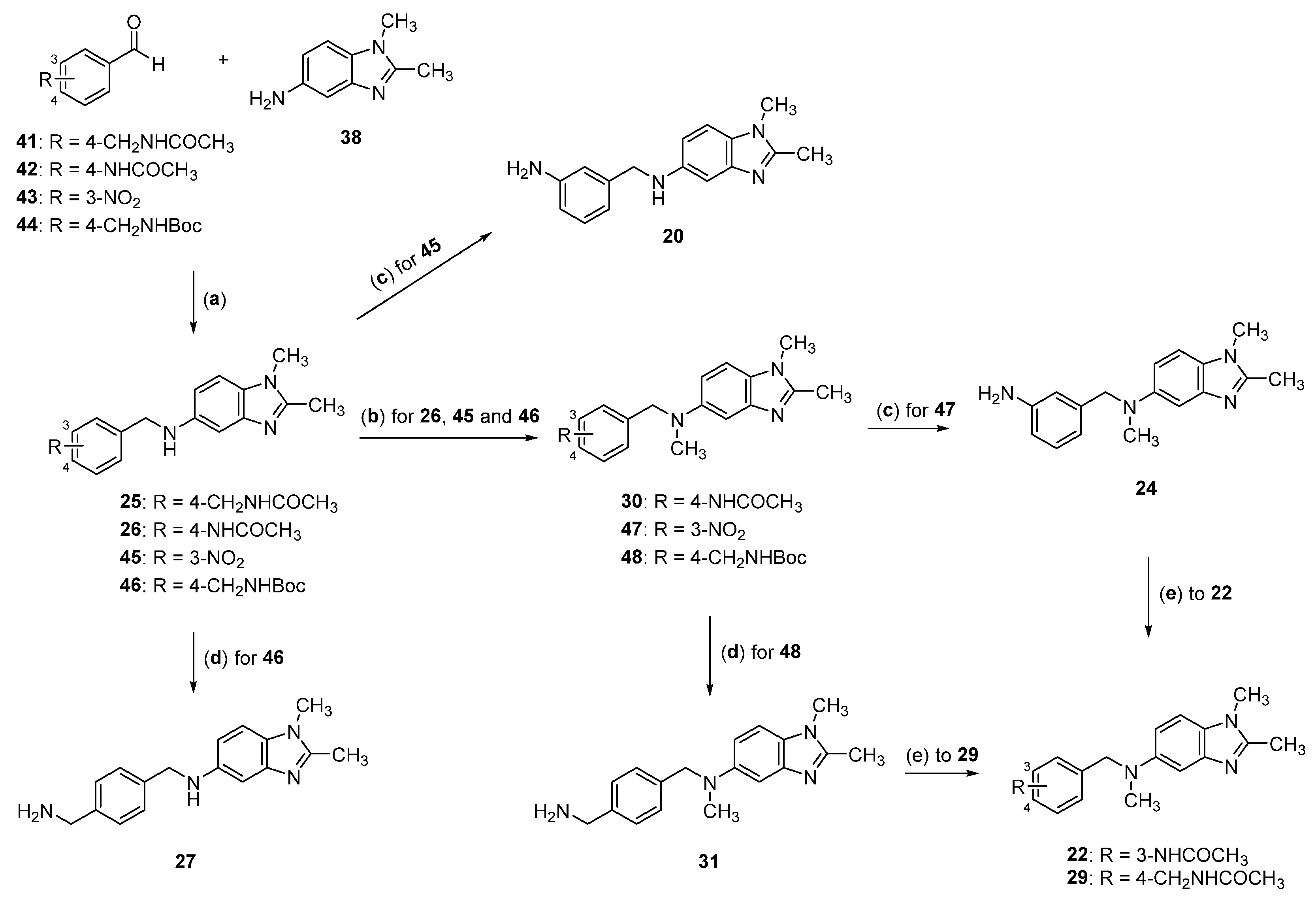
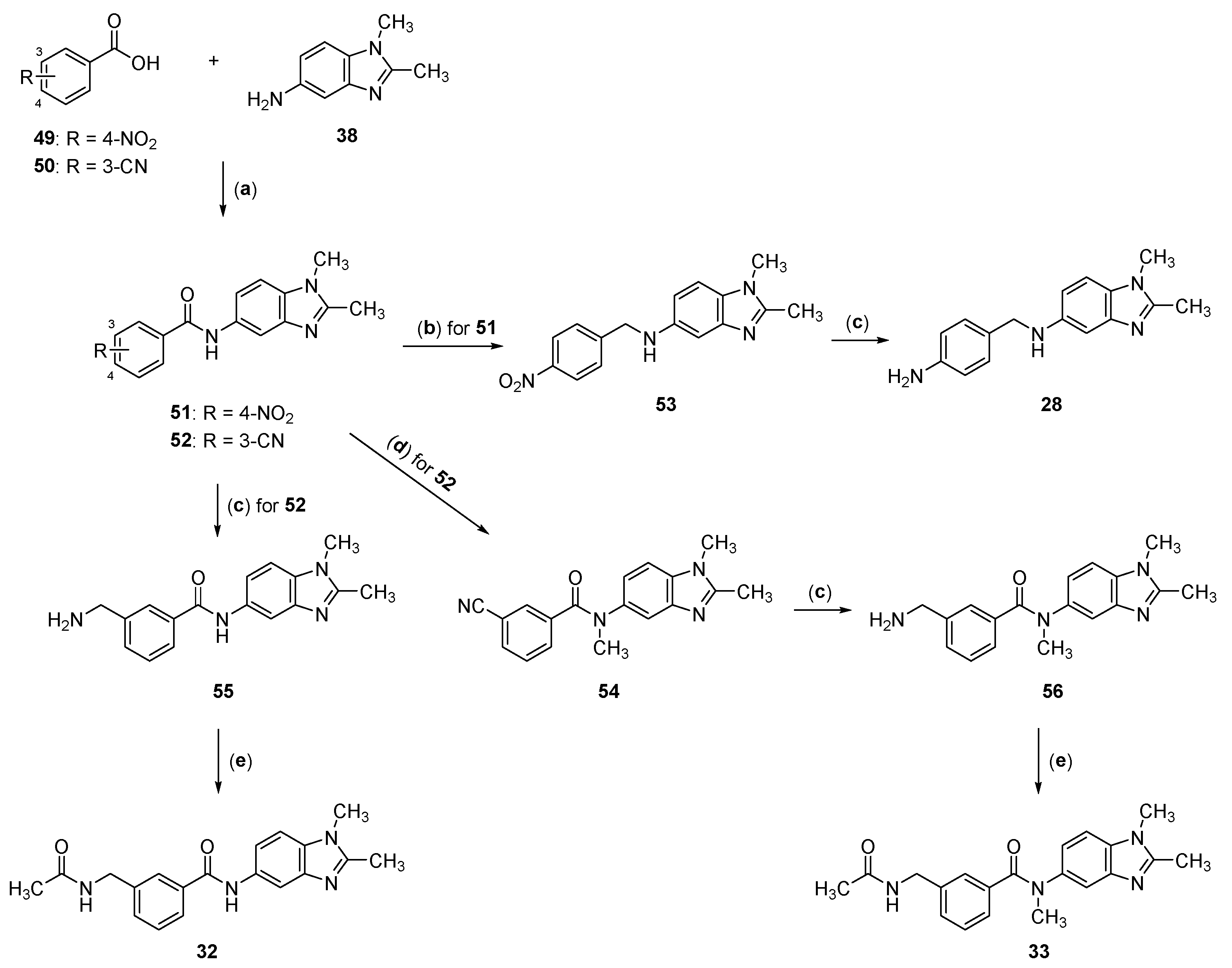
2.2. Synthesis of Compounds 17–33
2.3. Structure–Activity Relationship (SAR) Study
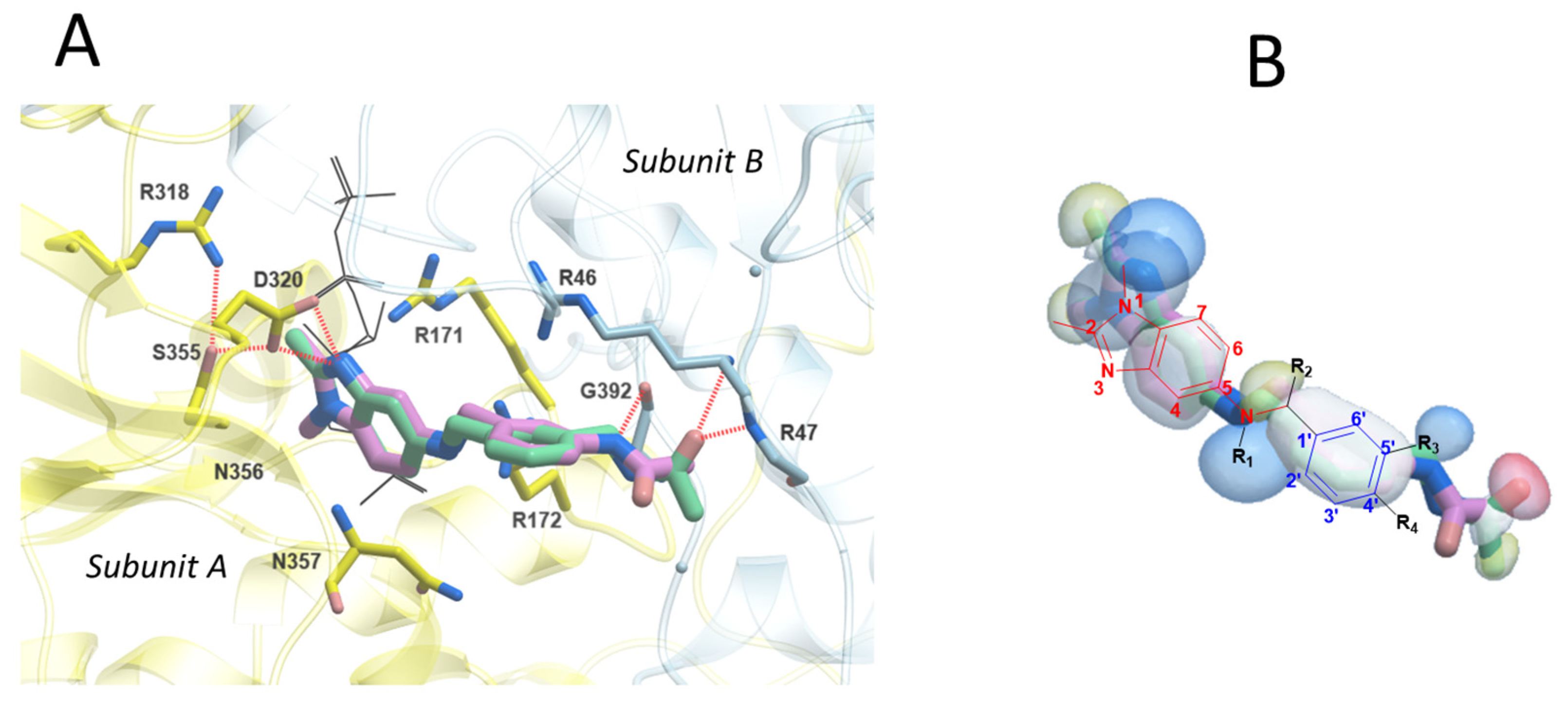
2.4. In Silico Analysis of Inhibitor and Activator Binding Pocket
2.5. Rationale for Modulators’ Activity
2.6. In Vitro Pharmacokinetic Studies
3. Materials and Methods
3.1. Chemistry
3.2. NAPRT Activity Screening Assay
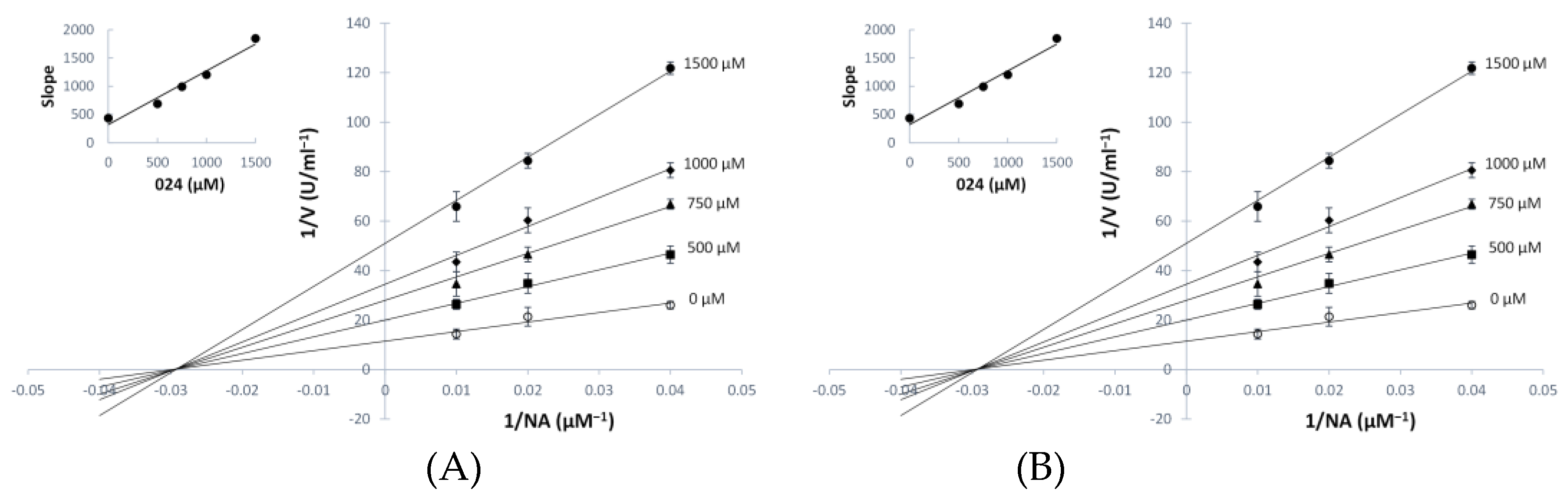
3.3. Kinetic Analyses
3.4. In Silico Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dölle, C.; Hvidsten Skoge, R.; VanLinden, M.R.; Ziegler, M. NAD biosynthesis in humans-enzymes, metabolites and therapeutic aspects. Curr. Top. Med. Chem. 2013, 13, 2907–2917. [Google Scholar] [CrossRef] [Green Version]
- Zapata-Pérez, R.; Wanders, R.J.A.; Karnebeek, C.D.M.; Houtkooper, R.H. NAD+ Homeostasis in Human Health and Disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef]
- Pankiewicz, K.W.; Petrelli, R.; Singh, R.; Felczak, K. Nicotinamide adenine dinucleotide based therapeutics, update. Curr. Med. Chem. 2015, 22, 3991–4028. [Google Scholar] [CrossRef]
- Zamporlini, F.; Ruggieri, S.; Mazzola, F.; Amici, A.; Orsomando, G.; Raffaelli, N. Novel assay for simultaneous measurement of pyridine mononucleotides synthesizing activities allows dissection of the NAD(+) biosynthetic machinery in mammalian cells. FEBS J. 2014, 281, 5104–5119. [Google Scholar] [CrossRef]
- Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- Goldinger, S.M.; Gobbi Bischof, S.; Fink-Puches, R.; Klemke, C.-D.; Dréno, B.; Bagot, M.; Dummer, R. Efficacy and safety of APO866 in patients with refractory or relapsed cutaneous T-cell lymphoma: A phase 2 clinical trial. JAMA Dermatol. 2016, 152, 837–839. [Google Scholar] [CrossRef] [Green Version]
- Von Heideman, A.; Berglund, A.; Larsson, R.; Nygren, P. Safety and Efficacy of NAD depleting cancer drugs: Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.-R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors, and DNA repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Q.; Lei, J.; Mao, L.H.; Wang, Q.L.; Xu, F.; Ran, T.; Zhou, Z.H.; He, S. NAMPT and NAPRT, key enzymes in NAD salvage synthesis pathway, are of negative prognostic value in colorectal cancer. Front. Oncol. 2019, 9, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent advances in NAMPT inhibitors: A novel immunotherapic strategy. Front. Pharmacol. 2020, 12, 656. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two metabolic enzymes with key roles in inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaut, Z.N.; Solomon, H.M. Inhibition of nicotinate phosphoribosyltransferase in human platelet lysate by nicotinic acid analogs. Biochem. Pharmacol. 1971, 20, 2903–2906. [Google Scholar] [CrossRef] [PubMed]
- Gaut, Z.N.; Solomon, H.M. Inhibition of nicotinate phosphoribosyl transferase by nonsteroidal anti-inflammatory drugs: A possible mechanism of action. J. Pharm. Sci. 1971, 60, 1887–1888. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Piacente, F.; Walter, M.; Fratta, S.; Ghanem, M.; Benzi, A.; Caffa, I.; Kurkin, A.V.; Altieri, A.; Herr, P.; et al. Structure-based identification and biological characterization of new NAPRT inhibitors. Pharmaceuticals 2022, 15, 855. [Google Scholar] [CrossRef]
- Ghanem, M.S.; Caffa, I.; Del Rio, A.; Franco, J.; Parenti, M.D.; Monacelli, F.; Cea, M.; Khalifa, A.; Nahimana, A.; Duchosal, M.A.; et al. Identification of NAPRT inhibitors with anti-cancer properties by in silico drug discovery. Pharmaceuticals 2022, 15, 848. [Google Scholar] [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]
- Zhang, M.; Ying, W. NAD+ deficiency is a common central pathological factor of a number of diseases and aging: Mechanisms and therapeutic implications. Antioxid. Redox Signal. 2019, 30, 890−900. [Google Scholar] [CrossRef]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Hampton Sessions, E.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD+ with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 3241. [Google Scholar] [CrossRef] [Green Version]
- Pinkerton, A.B.; Sessions, E.H.; Hershberger, P.; Maloney, P.R.; Peddibhotla, S.; Hopf, M.; Sergienko, E.; Ma, C.T.; Smith, L.H.; Jackson, M.R.; et al. Optimization of a urea-containing series of nicotinamide phosphoribosyltransferase (NAMPT) activators. Bioorg. Med. Chem. Lett. 2021, 41, 128007. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [Green Version]
- Reid, R.C.; Hansford, K.; Stoermer, M.J.; McGeary, R.P.; Fairlie, D.P.; Schafer, K. Derivatives Compounds and Inhibitors of Phospholipases. Patent WO0208189Al, 31 January 2002. [Google Scholar]
- Herre, S.; Steinle, W.; Rück-Braun, K. Synthesis of Photoswitchable hemithioindigo-based ω-amino acids and application in boc-based peptide assembly. Synthesis 2005, 2005, 3297–3300. [Google Scholar]
- Xi, N.; Li, M.; Peng, J.; Li, X.; Zhang, T.; Hu, H.; Chen, W.; Bai, C.; Ke, D.; Chen, P. Substituted Heteroaryl Compounds and Methods of. use. Patent WO201999311A1, 05 2019. [Google Scholar]
- Obreza, A.; Stegnar, M.; Urleb, U. Novel non-covalent azaphenylalanine thrombin inhibitors with an aminomethyl or amino group at the P1 position. Pharmazie 2004, 59, 659–667. [Google Scholar] [CrossRef]
- Galassi, L.; Di Stefano, M.; Brunetti, L.; Orsomando, G.; Amici, A.; Ruggieri, S.; Magni, G. Characterization of human nicotinate phosphoribosyltransferase: Kinetic studies, structure prediction and functional analysis by site-directed mutagenesis. Biochimie 2012, 94, 300–309. [Google Scholar] [CrossRef]
- Duarte-Pereira, S.; Fajarda, O.; Matos, S.; Luís Oliveira, J.; Silva, R.M. NAPRT Expression Regulation Mechanisms: Novel Functions Predicted by a Bioinformatics Approach. Genes 2021, 12, 2022. [Google Scholar] [CrossRef] [PubMed]
- Micheli, F.; Bacchi, A.; Braggio, S.; Castelletti, L.; Cavallini, P.; Cavanni, P.; Cremonesi, S.; Dal Cin, M.; Feriani, A.; Gehanne, S.; et al. 1,2,4-Triazolyl 5-azaspiro[2.4]heptanes: Lead identification and early lead optimization of a new series of potent and selective dopamine D3 receptor antagonists. J. Med. Chem. 2016, 59, 8549–8576. [Google Scholar] [CrossRef] [PubMed]
- Del Bello, F.; Bonifazi, A.; Giorgioni, G.; Cifani, C.; Micioni Di Bonaventura, M.V.; Petrelli, R.; Piergentili, A.; Fontana, S.; Mammoli, V.; Yano, H.; et al. 1-[3-(4-Butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28-1) as a model for the rational design of a novel class of brain penetrant ligands with high affinity and selectivity for dopamine D4 receptor. J. Med. Chem. 2018, 61, 3712–3725. [Google Scholar] [CrossRef]
- Del Bello, F.; Farande, A.; Giannella, M.; Piergentili, A.; Quaglia, W.; Benicchi, T.; Cappelli, F.; Nencini, A.; Salerno, M.; Thomas, R.J.; et al. Identification of 2-aminopyrimidine derivatives as inhibitors of the canonical Wnt signalling pathway. Bioorg. Med. Chem. 2015, 23, 5725–5733. [Google Scholar] [CrossRef]
- Summerfield, S.G.; Jeffrey, P. In vitro prediction of brain penetration—A case for free thinking? Expert Opin. Drug Discov. 2006, 6, 595–607. [Google Scholar] [CrossRef]
- Del Bello, F.; Bonifazi, A.; Giorgioni, G.; Piergentili, A.; Sabbieti, M.G.; Agas, D.; Dell’Aera, M.; Matucci, R.; Górecki, M.; Pescitelli, G.; et al. Novel Potent muscarinic receptor antagonists: Investigation on the nature of lipophilic substituents in the 5- and/or 6-positions of the 1,4-dioxane nucleus. J. Med. Chem. 2020, 63, 5763–5782. [Google Scholar] [CrossRef]
- Pavletić, P.; Semeano, A.; Yano, H.; Bonifazi, A.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; Sabbieti, M.G.; Agas, D.; Santoni, G.; et al. Highly potent and selective dopamine D4 receptor antagonists potentially useful for the treatment of glioblastoma. J. Med. Chem. 2022, 65, 12124–12139. [Google Scholar] [CrossRef]
- Minazzato, G.; Marangoni, E.; Fortunato, C.; Petrelli, R.; Cappellacci, L.; Del Bello, F.; Sorci, L.; Gasparrini, M.; Piacente, F.; Bruzzone, S.; et al. A Versatile Continuous Fluorometric Enzymatic Assay for Targeting Nicotinate Phosphoribosyltransferase. Molecules 2023, 28, 961. [Google Scholar] [CrossRef]
- Sorci, L.; Martynowski, D.; Rodionov, D.A.; Eyobo, Y.; Zogaj, X.; Klose, K.E.; Nikolaev, E.V.; Magni, G.; Zhang, H.; Osterman, A.L. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc. Natl. Acad. Sci. USA 2009, 106, 3083–3088. [Google Scholar] [CrossRef] [Green Version]
- Segel, I. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley and Sons Inc.: New York, NY, USA, 1993; pp. 1–957. [Google Scholar]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Totrov, M. Atomic property fields: Generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem. Biol. Drug Des. 2008, 71, 5–27. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | % Inhibition | -Fold Stimulation |
|---|---|---|---|
| 17 |  | 30.3 ± 6.0 | -b |
| 18 |  | 46.1 ± 8.1 | - |
| 19 |  | - | - |
| 20 |  | - | 1.25 ± 0.05 |
| 21 |  | - | - |
| 22 |  | - | - |
| 23 |  | - | 1.14 ± 0.05 |
| 24 |  | - | 1.44 ± 0.04 |
| 25 |  | 13.0 ± 3.1 | - |
| 26 |  | - | - |
| 27 |  | - | - |
| 28 |  | - | - |
| 29 |  | 12.0 ± 2.2 | - |
| 30 |  | - | - |
| 31 |  | - | 1.58 ± 0.07 |
| 32 |  | - | 1.47 ± 0.07 |
| 33 |  | - | - |
| 18 | ||
|---|---|---|
| Kinetic Solubility (pH 7.4) | µM | 312 |
| mg/mL | 0.1302 | |
| Protein Binding a | Human plasma (FU) b | 14.6 |
| Mouse plasma (FU) b | >50 | |
| Hepatic Intrinsic Clearance (CLi) in Liver Microsomes | Human Cli (µL/min/mg protein) | <9.92 |
| WSM c in vivo prediction (mL/min/kg) | 6.83 | |
| Mouse Cli (µL/min/mg protein) | 120 | |
| WSM c in vivo prediction (mL/min/kg) | 26.9 | |
| Permeability d | Papp A-B MDCKII (nm/sec) | 38.3 |
| Papp B-A MDCKII (nm/sec) | 65.9 | |
| Efflux Ratio MDCKII (B-A/A-B) | 1.7 | |
| Papp A-B MDCKII-MDR1 (nm/sec) | 3.39 | |
| Papp B-A MDCKII-MDR1 (nm/sec) | 174 | |
| Efflux Ratio MDCKII-MDR1 (B-A/A-B) | 51.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldassarri, C.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; Fontana, S.; Mammoli, V.; Minazzato, G.; Marangoni, E.; Gasparrini, M.; Sorci, L.; et al. Properly Substituted Benzimidazoles as a New Promising Class of Nicotinate Phosphoribosyltransferase (NAPRT) Modulators. Pharmaceuticals 2023, 16, 189. https://doi.org/10.3390/ph16020189
Baldassarri C, Giorgioni G, Piergentili A, Quaglia W, Fontana S, Mammoli V, Minazzato G, Marangoni E, Gasparrini M, Sorci L, et al. Properly Substituted Benzimidazoles as a New Promising Class of Nicotinate Phosphoribosyltransferase (NAPRT) Modulators. Pharmaceuticals. 2023; 16(2):189. https://doi.org/10.3390/ph16020189
Chicago/Turabian StyleBaldassarri, Cecilia, Gianfabio Giorgioni, Alessandro Piergentili, Wilma Quaglia, Stefano Fontana, Valerio Mammoli, Gabriele Minazzato, Elisa Marangoni, Massimiliano Gasparrini, Leonardo Sorci, and et al. 2023. "Properly Substituted Benzimidazoles as a New Promising Class of Nicotinate Phosphoribosyltransferase (NAPRT) Modulators" Pharmaceuticals 16, no. 2: 189. https://doi.org/10.3390/ph16020189