
“Dual Anta-Inhibitors” of the A2A Adenosine Receptor and Casein Kinase CK1delta: Synthesis, Biological Evaluation, and Molecular Modeling Studies
 , , , , ,
, , , , ,  , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
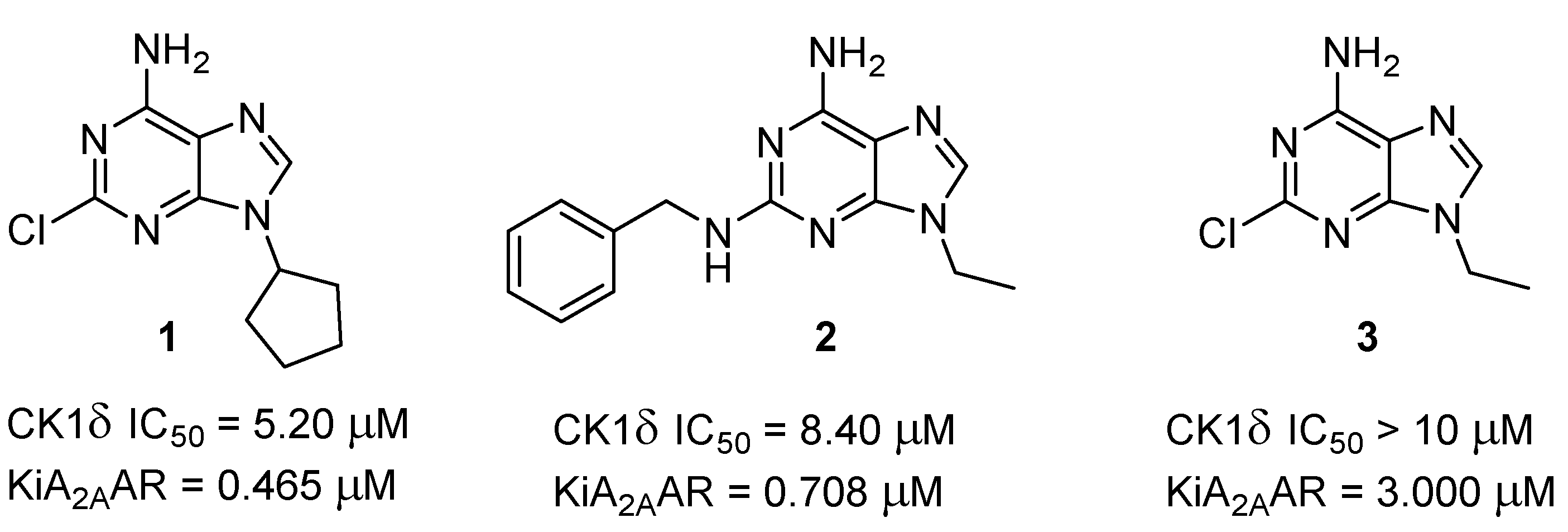
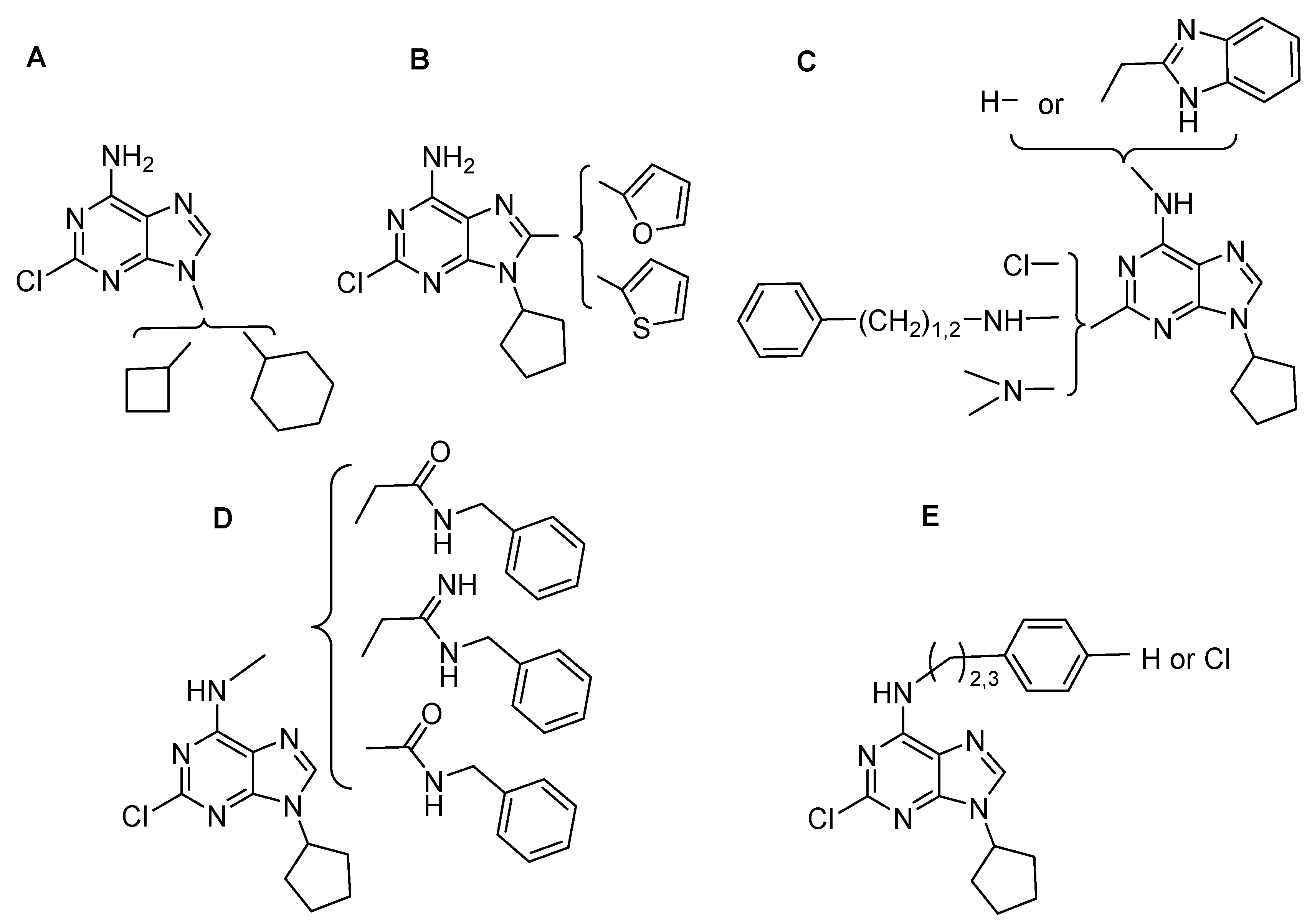
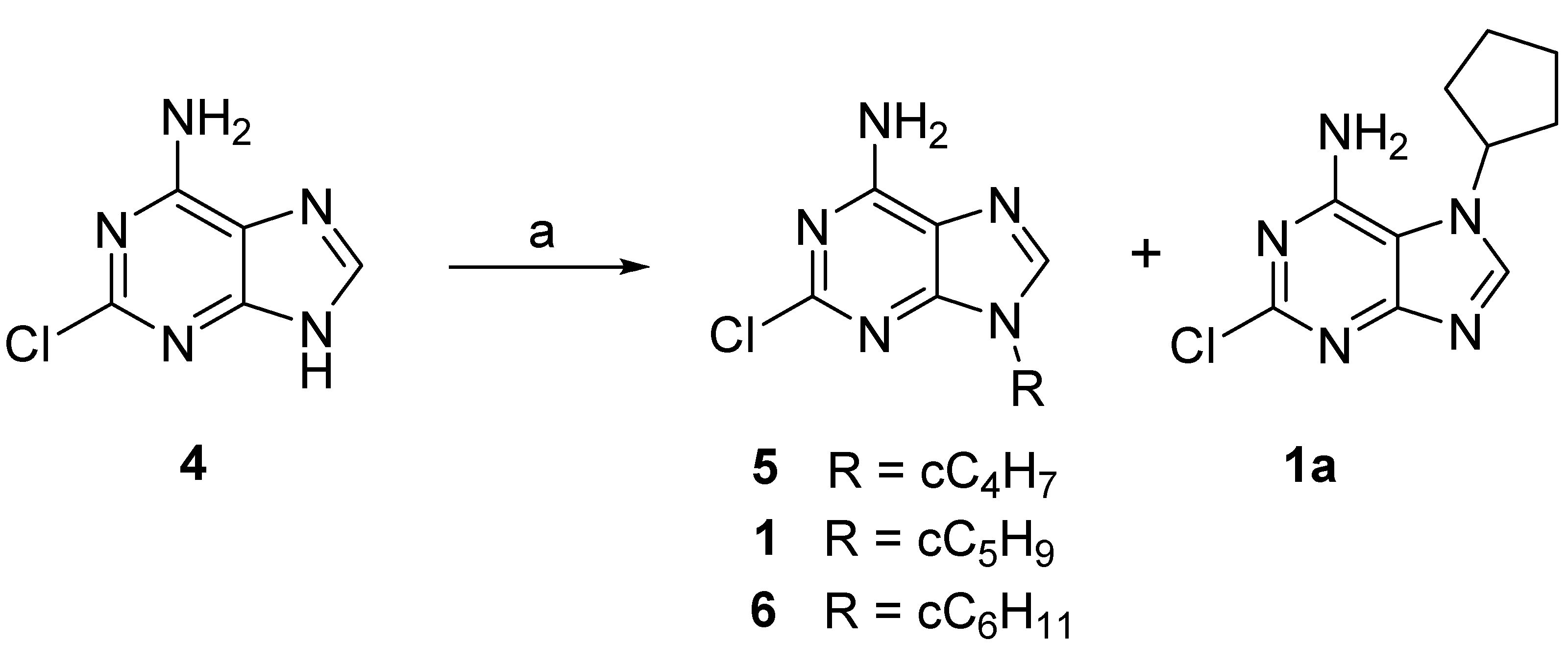
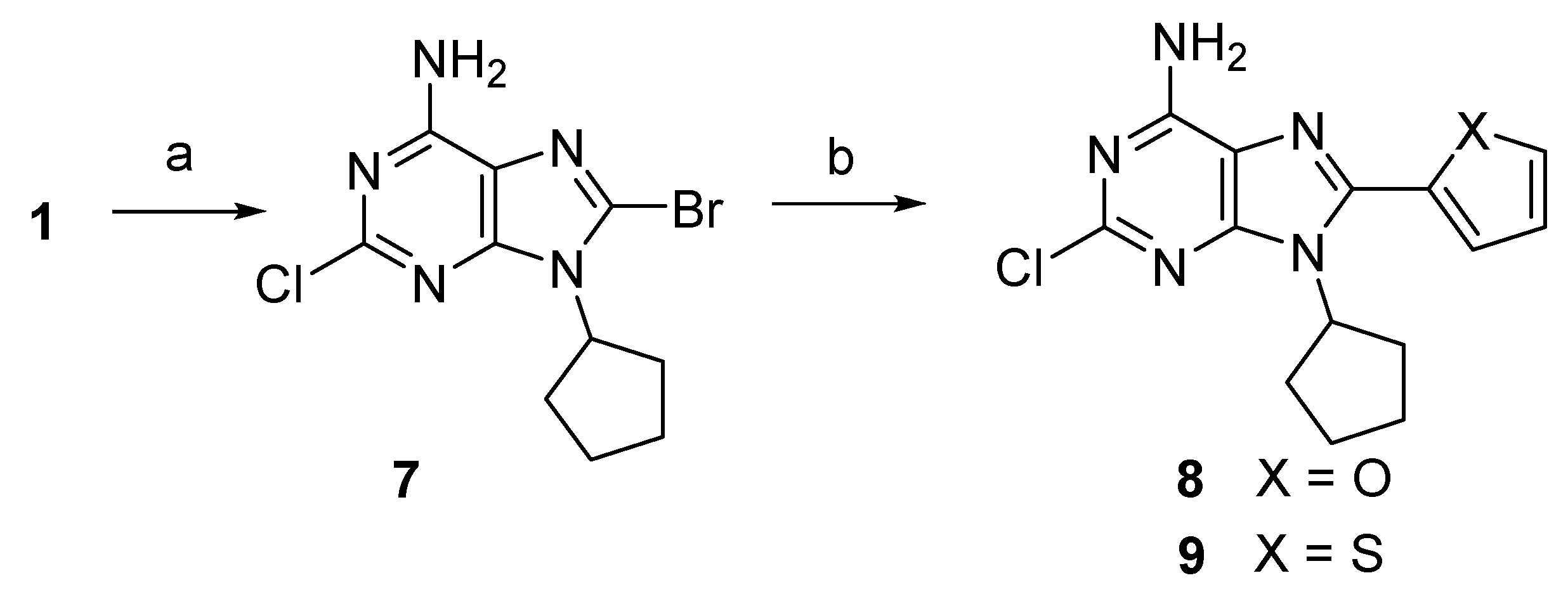
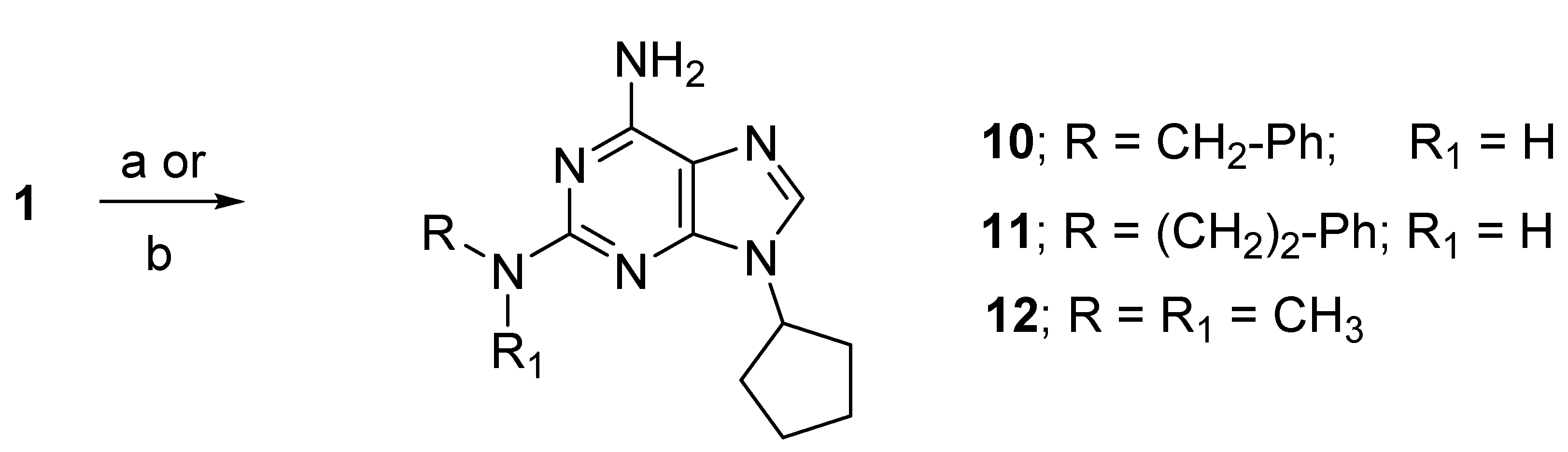
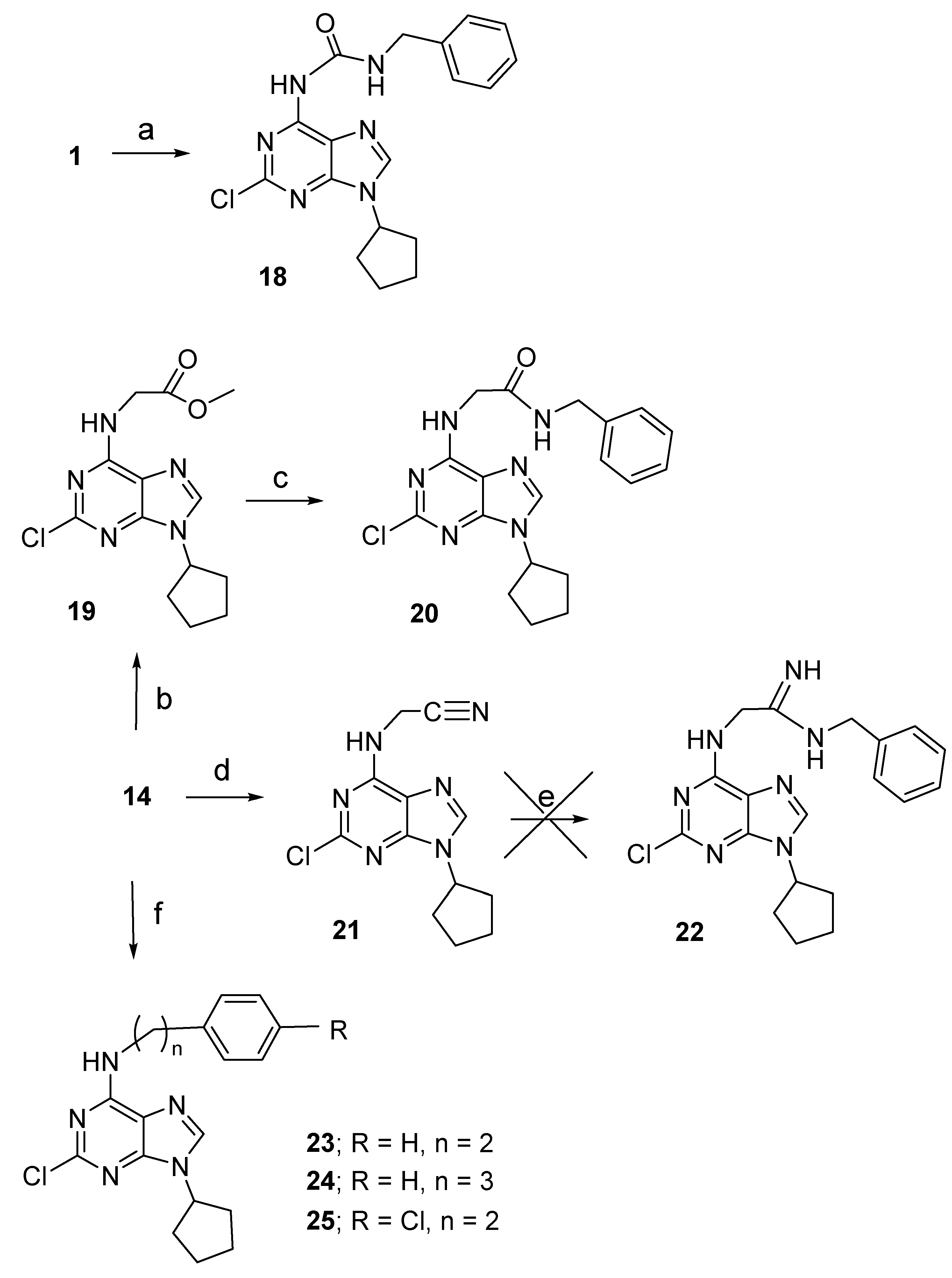
2.1. Chemistry
2.2. CK1δ Inhibitory Activity Assay
2.3. Binding Assay at A1, A2A, and A3 ARs and Functional Studies at A2BARs
2.4. Functional Activity at Human A2AAR
2.5. Computational Studies
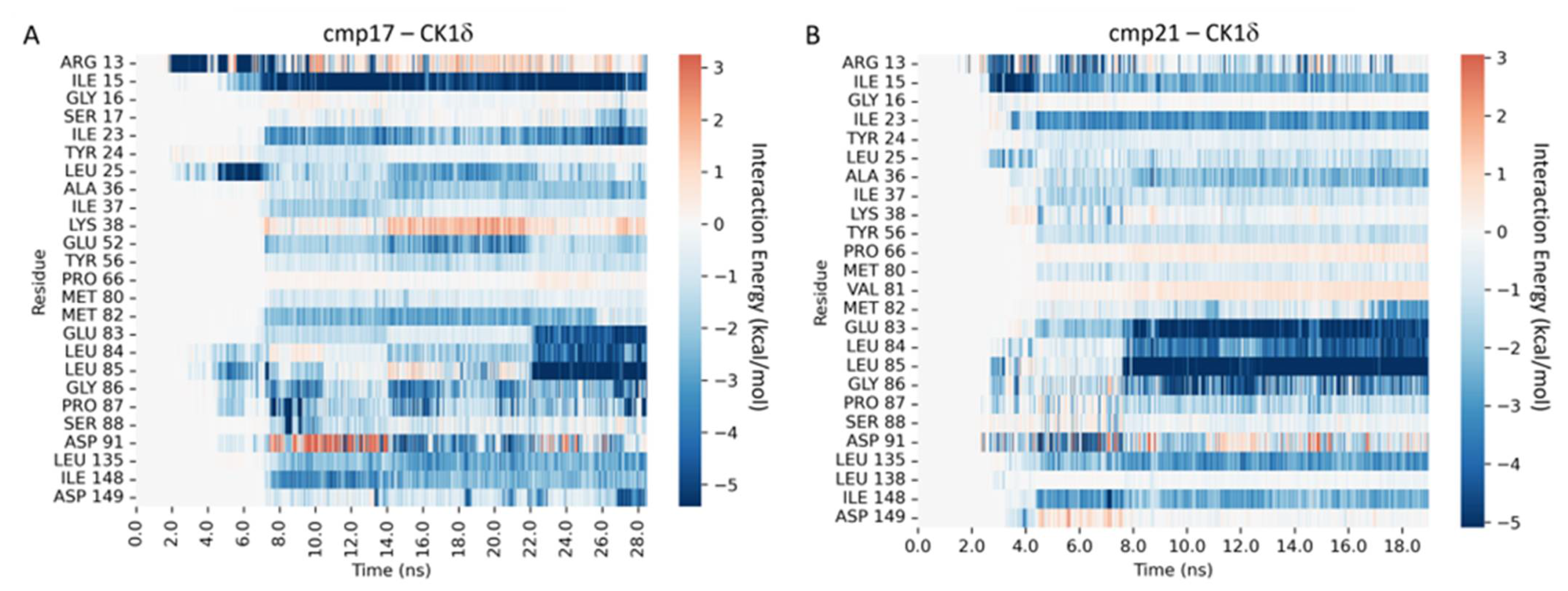
2.5.1. CK1δ Inhibition
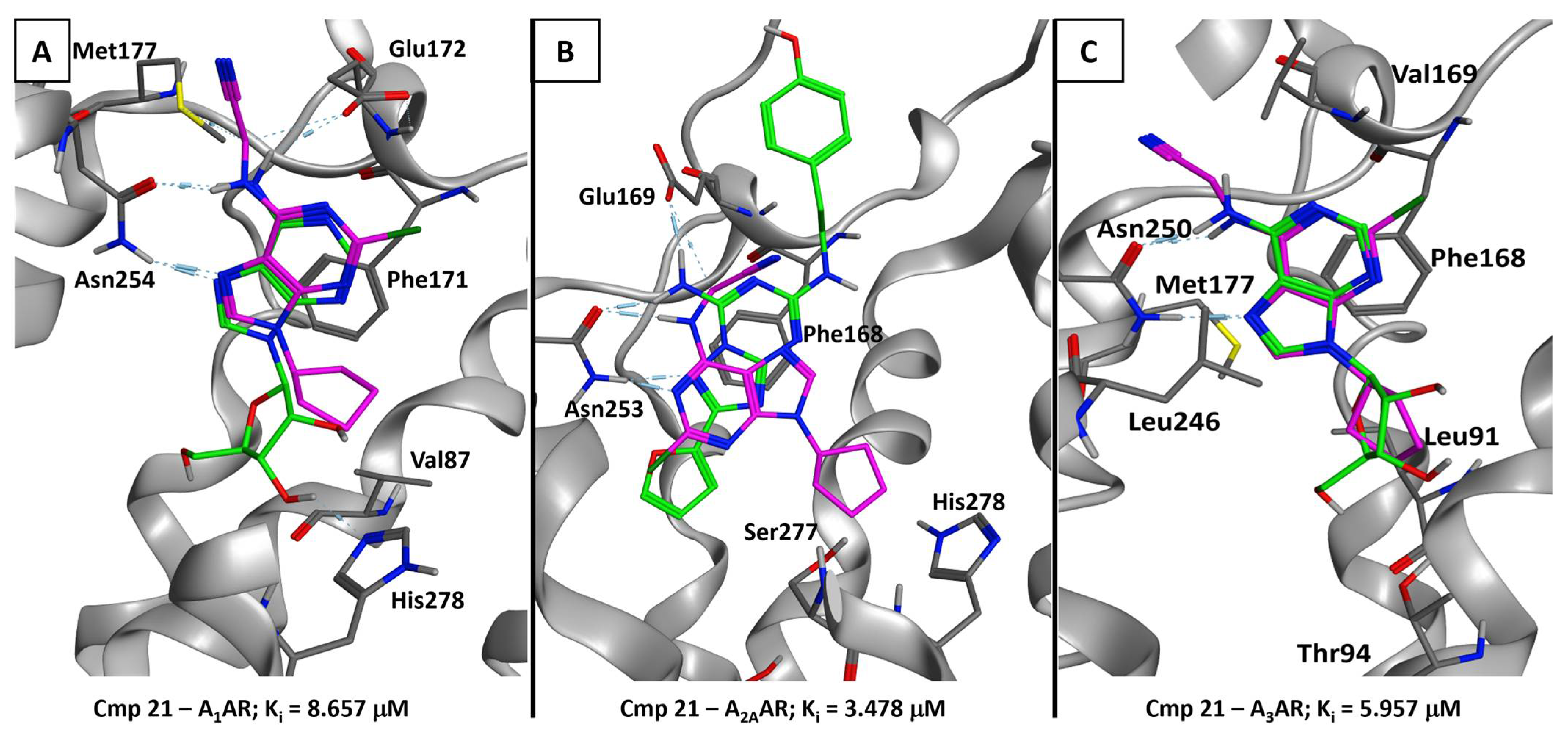
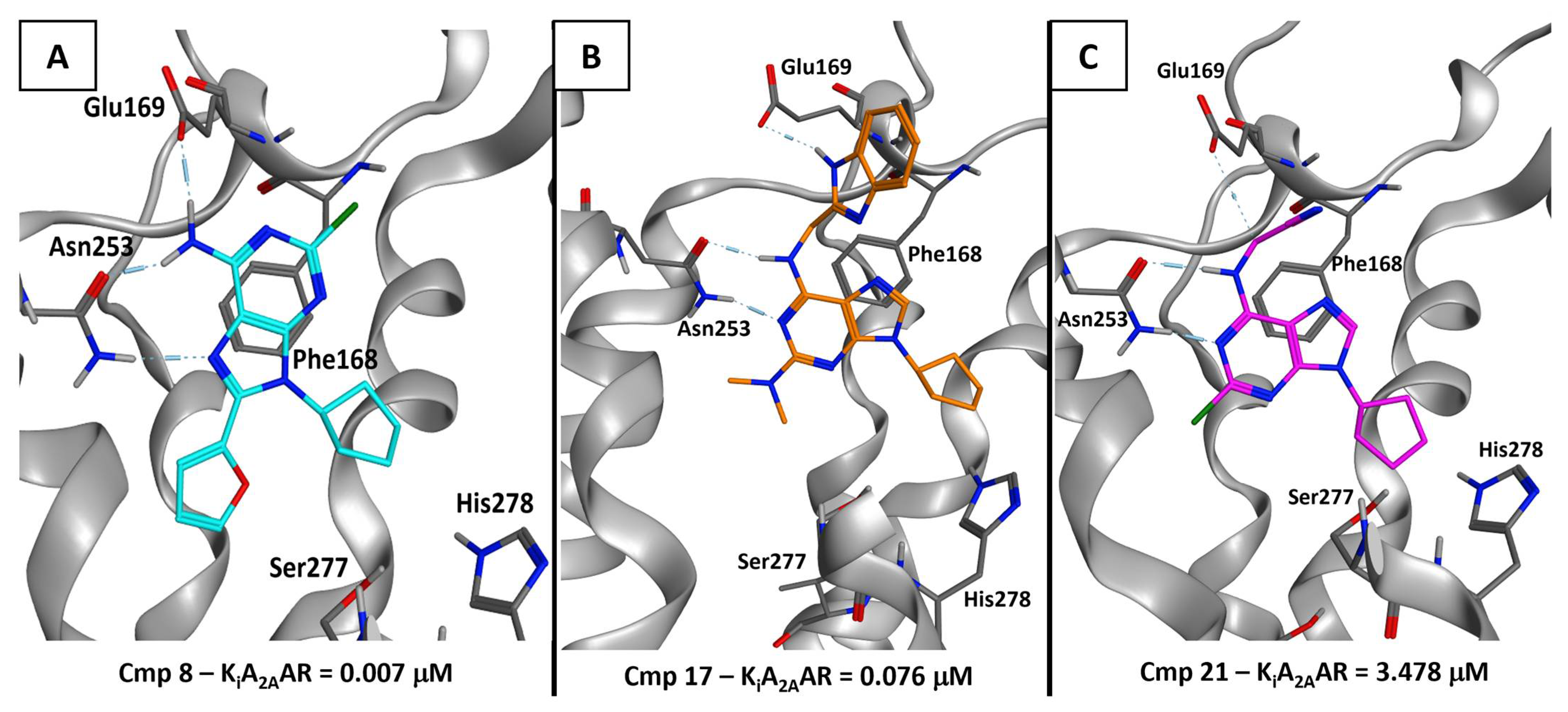
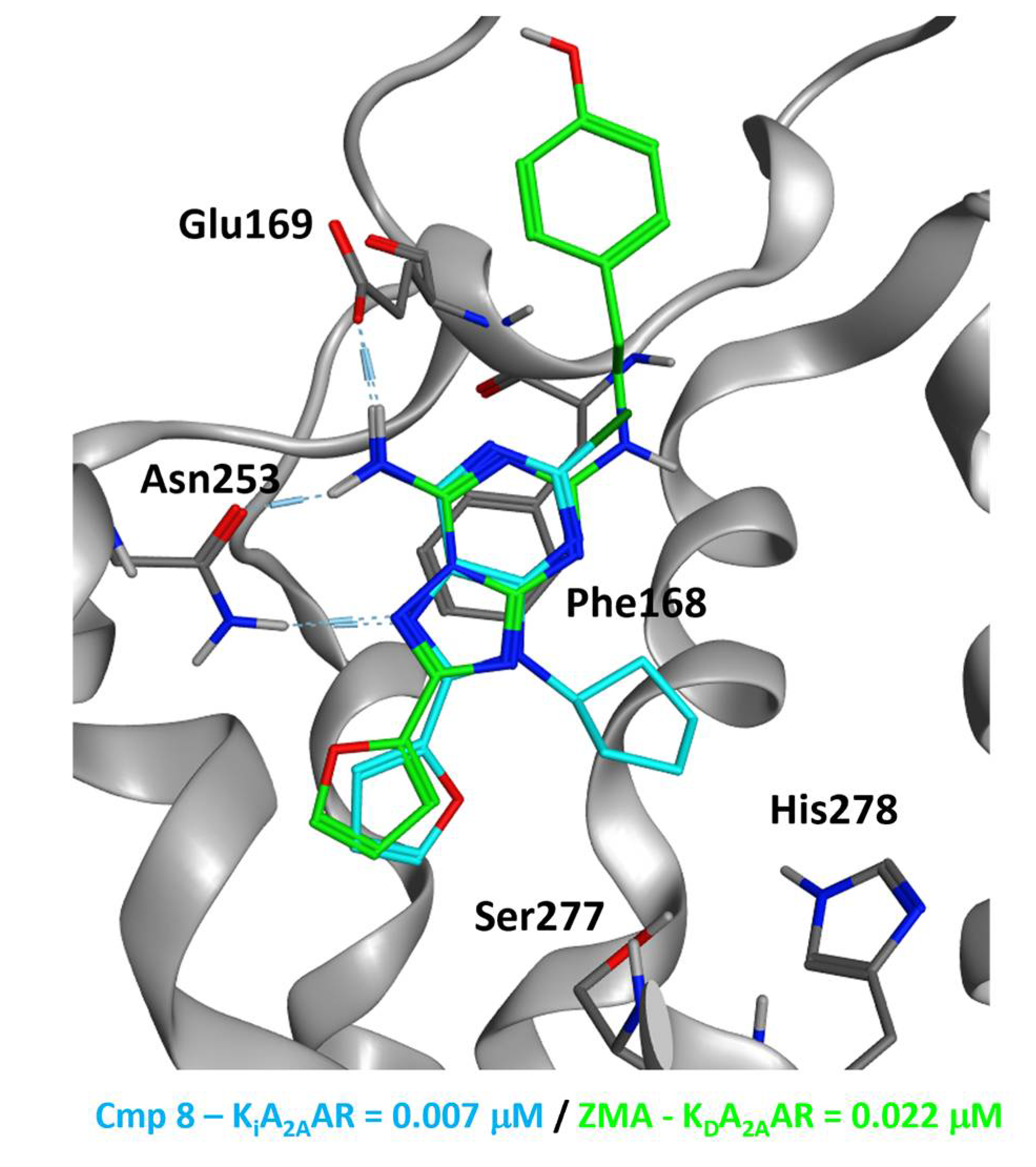
2.5.2. Adenosine Receptor Inhibition
2.5.3. Supervised Molecular Dynamics (SuMD) Simulations
3. Materials and Methods
3.1. Chemical Synthesis
General Methods
3.2. CK1δ Activity Assays
3.3. Biological Assays at Human Adenosine Receptors
3.3.1. Cell Culture
3.3.2. Membrane Preparation
3.3.3. Binding Assay
3.3.4. Functional Study at Human A2A and A2B Ars
3.3.5. Statistical Analysis
3.4. Molecular Modeling Studies
3.4.1. Software Overview
3.4.2. Protein Preparation
3.4.3. Ligand Preparation
3.4.4. Molecular Docking
3.4.5. System Setup for Supervised Molecular Dynamics (SuMD) Simulations
3.4.6. Supervised Molecular Dynamics (SuMD) Simulations
3.4.7. SuMD Trajectories Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheong, J.K.; Virshup, D.M. Casein kinase 1: Complexity in the family. Int. J. Biochem. Cell Biol. 2011, 43, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Yang, H.; Feng, F.; Liu, W.; Chen, Y.; Sun, H. Achieving effective and selective CK1 inhibitors through structure modification. Future Med. Chem. 2021, 13, 505–528. [Google Scholar] [CrossRef]
- Gross, S.D.; Anderson, R.A. Casein kinase I: Spatial organization and positioning of a multifunctional protein kinase family. Cell. Signal. 1998, 10, 699–711. [Google Scholar] [CrossRef]
- Xu, P.; Ianes, C.; Gartner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 2019, 715, 144005. [Google Scholar] [CrossRef]
- Catarzi, D.; Varano, F.; Vigiani, E.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Colotta, V. Casein Kinase 1 delta Inhibitors as Promising Therapeutic Agents for Neurodegenerative Disorders. Curr. Med. Chem. 2022, 29, 4698–4737. [Google Scholar] [CrossRef]
- Li, S.S.; Dong, Y.H.; Liu, Z.P. Recent Advances in the Development of Casein Kinase 1 Inhibitors. Curr. Med. Chem. 2021, 28, 1585–1604. [Google Scholar] [CrossRef]
- Oumata, N.; Bettayeb, K.; Ferandin, Y.; Demange, L.; Lopez-Giral, A.; Goddard, M.L.; Myrianthopoulos, V.; Mikros, E.; Flajolet, M.; Greengard, P.; et al. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 2008, 51, 5229–5242. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyi, A.; Nilchan, N.; Quereda, V.; Noguchi, Y.; Ruiz, C.; Grant, W.; Cameron, M.; Duckett, D.; Roush, W. Development of dual casein kinase 1 delta/1epsilon (CK1delta/epsilon) inhibitors for treatment of breast cancer. Bioorg. Med. Chem. 2018, 26, 590–602. [Google Scholar] [CrossRef]
- Bibian, M.; Rahaim, R.J.; Choi, J.Y.; Noguchi, Y.; Schurer, S.; Chen, W.; Nakanishi, S.; Licht, K.; Rosenberg, L.H.; Li, L.; et al. Development of highly selective casein kinase 1delta/1epsilon (CK1delta/epsilon) inhibitors with potent antiproliferative properties. Bioorg. Med. Chem. Lett. 2013, 23, 4374–4380. [Google Scholar] [CrossRef] [Green Version]
- Roush, W.R.; Ayad, N.; Rahaim, R.; Simanski, S.P.; Bibian, M. WEE1 degradation inhibitors. WO2013130461A1 2013. [Google Scholar]
- Rosenberg, L.H.; Lafitte, M.; Quereda, V.; Grant, W.; Chen, W.; Bibian, M.; Noguchi, Y.; Fallahi, M.; Yang, C.; Chang, J.C.; et al. Therapeutic targeting of casein kinase 1 delta in breast cancer. Sci. Transl. Med. 2015, 7, 318ra202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IJzerman, A.P.; Jacobson, K.A.; Muller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef] [PubMed]
- Marti Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, C.; Marucci, G.; Catarzi, D.; Colotta, V.; Francucci, B.; Spinaci, A.; Varano, F.; Volpini, R. A2A Adenosine Receptor Antagonists and their Potential in Neurological Disorders. Curr. Med. Chem. 2022, 29, 4780–4795. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; IJzerman, A.P.; Muller, C.E. Medicinal chemistry of P2 and adenosine receptors: Common scaffolds adapted for multiple targets. Biochem. Pharmacol. 2021, 187, 114311. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Navarro, G. Adenosine A2A Receptor Antagonists in Neurodegenerative Diseases: Huge Potential and Huge Challenges. Front. Psychiatry 2018, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, A.; Chen, J.F.; Uchida, S.; Durlach, C.; King, S.M.; Jenner, P. The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease. Molecules 2022, 27, 2366. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic. Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Ben, D.D.; Lambertucci, C.; Marti Navia, A.; Spinaci, A.; Volpini, R.; Buccioni, M. Combined therapy of A1AR agonists and A2AAR antagonists in neuroinflammation. Molecules 2021, 26, 1188. [Google Scholar] [CrossRef]
- Ferrante, A.; De Simone, R.; Ajmone-Cat, M.A.; Minghetti, L.; Popoli, P. Adenosine receptors and neuroinflammation. In The Adenosine Receptors; Borea, P., Varani, K., Gessi, S., Merighi, S., Vincenzi, F., Eds.; The Receptors; Humana Press: Cham, Switzerland, 2018; Volume 34. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Pinna, A.; Tronci, E.; Schintu, N.; Simola, N.; Volpini, R.; Pontis, S.; Cristalli, G.; Morelli, M. A new ethyladenine antagonist of adenosine A2A receptors: Behavioral and biochemical characterization as an antiparkinsonian drug. Neuropharmacology 2010, 58, 613–623. [Google Scholar] [CrossRef]
- Yu, F.; Zhu, C.; Xie, Q.; Wang, Y. Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem. 2020, 63, 12196–12212. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, C.; Spinaci, A.; Buccioni, M.; Dal Ben, D.; Ngouadjeu Ngnintedem, M.A.; Kachler, S.; Marucci, G.; Klotz, K.N.; Volpini, R. New A2A adenosine receptor antagonists: A structure-based upside-down interaction in the receptor cavity. Bioorg. Chem. 2019, 92, 103183. [Google Scholar] [CrossRef]
- Spinaci, A.; Lambertucci, C.; Buccioni, M.; Dal Ben, D.; Graiff, C.; Barbalace, M.C.; Hrelia, S.; Angeloni, C.; Tayebati, S.K.; Ubaldi, M.; et al. A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable? Molecules 2022, 27, 2386. [Google Scholar] [CrossRef]
- Thomas, A.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Kachler, S.; Klotz, K.N.; Volpini, R. The Length and Flexibility of the 2-Substituent of 9-Ethyladenine Derivatives Modulate Affinity and Selectivity for the Human A2A Adenosine Receptor. ChemMedChem 2016, 11, 1829–1839. [Google Scholar] [CrossRef]
- Information, N.C.F.B. PubChem Compound Summary for CID 141174762, 2-Chloro-9-cyclopentylpurin-6-amine; National Library of Medicine (US): Bethesda, MD, USA, 2019. [Google Scholar]
- Lambertucci, C.; Vittori, S.; Mishra, R.C.; Dal Ben, D.; Klotz, K.N.; Volpini, R.; Cristalli, G. Synthesis and biological activity of trisubstituted adenines as A2A adenosine receptor antagonists. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1443–1446. [Google Scholar] [CrossRef]
- Lambertucci, C.; Buccioni, M.; Dal Ben, D.; Kachler, S.; Marucci, G.; Spinaci, A.; Thomas, A.; Klotz, K.N.; Volpini, R. New substituted 9-propyladenine derivatives as A2A adenosine receptor antagonists. Medchemcomm 2015, 6, 963–970. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Thomas, A.; Klotz, K.-N.; Federico, S.; Cacciari, B.; Spalluto, G.; Volpini, R. 8-(2-Furyl)adenine derivatives as A2A adenosine receptor ligands. Eur. J. Med. Chem. 2013, 70, 525–535. [Google Scholar] [CrossRef]
- Borrmann, T.; Abdelrahman, A.; Volpini, R.; Lambertucci, C.; Alksnis, E.; Gorzalka, S.; Knospe, M.; Schiedel, A.C.; Cristalli, G.; Müller, C.E. Structure-activity relationships of adenine and deazaadenine derivatives as ligands for adenine receptors, a new purinergic receptor family. J. Med. Chem. 2009, 52, 5974–5989. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, C.; Antonini, I.; Buccioni, M.; Dal Ben, D.; Kachare, D.D.; Volpini, R.; Klotz, K.N.; Cristalli, G. 8-Bromo-9-alkyl adenine derivatives as tools for developing new adenosine A2A and A2B receptors ligands. Bioorg. Med. Chem. 2009, 17, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.N.; Falgner, N.; Kachler, S.; Lambertucci, C.; Vittori, S.; Volpini, R.; Cristalli, G. [3H]HEMADO--a novel tritiated agonist selective for the human adenosine A3 receptor. Eur. J. Pharmacol. 2007, 556, 14–18. [Google Scholar] [CrossRef]
- Falsini, M.; Ceni, C.; Catarzi, D.; Varano, F.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Marti Navia, A.; Volpini, R.; Colotta, V. New 8-amino-1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives. Evaluation of different moieties on the 6-aryl ring to obtain potent and selective human A2A adenosine receptor antagonists. Bioorg. Med. Chem. Lett. 2020, 30, 127126. [Google Scholar] [CrossRef]
- Buccioni, M.; Marucci, G.; Dal Ben, D.; Giacobbe, D.; Lambertucci, C.; Soverchia, L.; Thomas, A.; Volpini, R.; Cristalli, G. Innovative functional cAMP assay for studying G protein-coupled receptors: Application to the pharmacological characterization of GPR17. Purinergic. Signal. 2011, 7, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Pavan, M.; Bassani, D.; Bolcato, G.; Bissaro, M.; Sturlese, M.; Moro, S. Computational Strategies to Identify New Drug Candidates against Neuroinflammation. Curr. Med. Chem. 2022, 29, 4756–4775. [Google Scholar] [CrossRef]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Implementing a Scoring Function Based on Interaction Fingerprint for Autogrow4: Protein Kinase CK1delta as a Case Study. Front. Mol. Biosci. 2022, 9, 909499. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Draper-Joyce, C.J.; Bhola, R.; Wang, J.; Bhattarai, A.; Nguyen, A.T.N.; Cowie-Kent, I.; O’Sullivan, K.; Chia, L.Y.; Venugopal, H.; Valant, C.; et al. Positive allosteric mechanisms of adenosine A(1) receptor-mediated analgesia. Nature 2021, 597, 571–576. [Google Scholar] [CrossRef]
- Batyuk, A.; Galli, L.; Ishchenko, A.; Han, G.W.; Gati, C.; Popov, P.A.; Lee, M.Y.; Stauch, B.; White, T.A.; Barty, A.; et al. Native phasing of X-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2016, 2, e1600292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margiotta, E.; Moro, S. A Comparison in the Use of the Crystallographic Structure of the Human A1 or the A2A Adenosine Receptors as a Template for the Construction of a Homology Model of the A3 Subtype. Appl. Sci. 2019, 9, 821. [Google Scholar] [CrossRef] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In International Workshop on Ant Colony Optimization and Swarm Intelligence. ANTS 2006; Dorigo, M., Gambardella, L.M., Birattari, M., Martinoli, A., Poli, R., Stützle, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 4150, pp. 247–258. [Google Scholar] [CrossRef]
- Bolcato, G.; Cescon, E.; Pavan, M.; Bissaro, M.; Bassani, D.; Federico, S.; Spalluto, G.; Sturlese, M.; Moro, S. A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1delta. Int. J. Mol. Sci. 2021, 22, 9741. [Google Scholar] [CrossRef]
- Albrecht-Kupper, B.E.; Leineweber, K.; Nell, P.G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic. Signal. 2012, 8 (Suppl. 1), 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.; Zhao, H.; Huang, X. Structural basis for the interaction between casein kinase 1 delta and a potent and selective inhibitor. J. Med. Chem. 2012, 55, 956–960. [Google Scholar] [CrossRef]
- Bolcato, G.; Bissaro, M.; Deganutti, G.; Sturlese, M.; Moro, S. New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations. Biomolecules 2020, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Bolcato, G.; Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Ribose and Non-Ribose A2A Adenosine Receptor Agonists: Do They Share the Same Receptor Recognition Mechanism? Biomedicines 2022, 10, 515. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2022, 179, 3496–3511. [Google Scholar] [CrossRef]
- Tosh, D.K.; Salmaso, V.; Campbell, R.G.; Rao, H.; Bitant, A.; Pottie, E.; Stove, C.P.; Liu, N.; Gavrilova, O.; Gao, Z.G.; et al. A3 adenosine receptor agonists containing dopamine moieties for enhanced interspecies affinity. Eur. J. Med. Chem. 2022, 228, 113983. [Google Scholar] [CrossRef]
- Cescon, E.; Bolcato, G.; Federico, S.; Bissaro, M.; Valentini, A.; Ferlin, M.G.; Spalluto, G.; Sturlese, M.; Moro, S. Scaffold Repurposing of in-House Chemical Library toward the Identification of New Casein Kinase 1 δinhibitors. ACS Med. Chem. Lett. 2020, 11, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Qualitative Estimation of Protein-Ligand Complex Stability through Thermal Titration Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 5715–5728. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Poucher, S.M.; Keddie, J.R.; Singh, P.; Stoggall, S.M.; Caulkett, P.W.; Jones, G.; Coll, M.G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2A selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332. J. Enzym. Inhib. Med. Chem. 2021, 36, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, M.; Bolcato, G.; Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Inspecting the Mechanism of Fragment Hits Binding on SARS-CoV-2 M(pro) by Using Supervised Molecular Dynamics (SuMD) Simulations. ChemMedChem 2021, 16, 2075–2081. [Google Scholar] [CrossRef]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, M.; Bolcato, G.; Deganutti, G.; Sturlese, M.; Moro, S. Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules 2019, 24, 2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755. [Google Scholar] [CrossRef]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase I epsilon induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730–738. [Google Scholar] [CrossRef]
- ULC, Chemical Computing Group. Molecular Operating Environment (MOE), Chemical Computing Group: Montreal, QC, Canada, 2019.01. 2019.
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory. Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W.; et al. Amber 10. University of California, San Francisco: San Francisco, CA, USA, 2008. [Google Scholar]
- Moro, S.; Deflorian, F.; Bacilieri, M.; Spalluto, G. Homology Modeling as Attractive Tool to Inspect GPCR Structural Plasticity. Curr. Pharm. Des. 2006, 12, 2175–2185. [Google Scholar] [CrossRef]
- QUACPAC 2.1.3.0: OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 15 December 2022).
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Bassani, D.; Pavan, M.; Sturlese, M.; Moro, S. Sodium or Not Sodium: Should Its Presence Affect the Accuracy of Pose Prediction in Docking GPCR Antagonists? Pharmaceuticals 2022, 15, 346. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [Green Version]
- Faller, R.; de Pablo, J.J. Constant pressure hybrid Molecular Dynamics–Monte Carlo simulations. J. Chem. Phys. 2002, 116, 7605. [Google Scholar] [CrossRef] [Green Version]
- Kräutler, V.; van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys 2020, 153, 04430. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Cmp | R1 | R2 | R3 | R4 | % Residual Activity at 40 μM ± sd | % Residual Activity at 10 μM ± sd | IC50 (μM) |
| 1 | Cl | cC5H9 | H | 0.70 ± 0.4 | 13 ± 7.4 | 5.20 ± 1.2 | |
| 5 | Cl | cC4H7 | H | 43 ± 3.2 | 37 ± 3.8 | 5.25 ± 0.9 | |
| 6 | Cl | cC6H11 | H | 30 ± 0.2 | 52 ± 3.0 | >10 | |
| 8 | Cl | cC5H9 | 2-furyl | 86 ± 1.6 | n.d. | >40 | |
| 9 | Cl | cC5H9 | 2-thienyl | 90 ± 4.6 | n.d. | >40 | |
| 10 | PhCH2NH | cC5H9 | H | 30 ± 4.1 | 48 ± 3.5 | 5.30 ± 1.0 | |
| 11 | Ph(CH2)2NH | cC5H9 | H | 88 ± 2.9 | n.d. | >40 | |
| 12 | (CH3)2N | cC5H9 | H | 19 ± 9.4 | 27 ± 4.0 | 1.75 ± 0.2 | |
| 15 | Cl | 22 ± 13 | 24 ± 14 | 1.53 ± 0.4 | |||
| 16 | PhCH2NH | 46 ± 31 | 26 ± 7.6 | 4.76 ± 0.3 | |||
| 17 | (CH3)2N | 22 ± 1.0 | 34 ± 5.0 | 0.59 ± 0.2 | |||
| 18 | CONHCH2Ph | 29 ± 3.9 | 38 ± 6.0 | 3.37 ± 2.5 | |||
| 19 | CH2COOCH3 | 44 ± 2.6 | 23 ± 2.4 | 2.55 ± 0.2 | |||
| 20 | CH2CONHCH2Ph | 29 ± 12 | 32 ± 16 | 1.87 ± 0.2 | |||
| 21 | CH2CN | 25 ± 9.6 | 13 ± 24 | 0.36 ± 0.1 | |||
| 23 | (CH2)2Ph | 14 ± 9.7 | 27 ± 20 | 0.66 ± 0.2 | |||
| 24 | (CH2)3Ph | 11 ± 19 | 26 ± 6.7 | 0.73 ± 0.1 | |||
| 25 | (CH2)2Ph-4Cl | 14 ± 4.3 | 31 ± 12 | 1.69 ± 1.0 | |||
| Cmp | R1 | R2 | R3 | R4 | KiA1 (μM) a | KiA2A (μM) b | KiA3 (μM) c | IC50 (μM) d |
|---|---|---|---|---|---|---|---|---|
| 1 | Cl | cC5H9 | H | 1.893 ± 0.160 | 0.465 ± 0.129 | 0.631 ± 0.062 | 5.20 | |
| 5 | Cl | cC4H7 | H | 1.533 ± 0.265 | 0.558 ± 0.151 | 7.224 ± 1874 | 5.25 | |
| 6 | Cl | cC6H11 | H | n.d. e | n.d. | n.d. | >10 | |
| 8 | Cl | cC5H9 | 2-furyl | 0.118 ± 0.010 | 0.007 ± 0.00001 | 0.140 ± 0.035 | >40 | |
| 9 | Cl | cC5H9 | 2-thienyl | 0.053 ± 0.009 | 0.036 ± 0.009 | 0.017 ± 0.002 | >40 | |
| 10 | PhCH2NH | cC5H9 | H | 3.957 ± 0.800 | 0.157 ± 0.040 | 0.298 ± 0.047 | 5.30 | |
| 11 | Ph(CH2)2NH | cC5H9 | H | 0.580 ± 0.046 | 0.012 ± 0.003 | 0.049 ± 0.008 | >40 | |
| 12 | (CH3)2N | cC5H9 | H | 3.339 ± 0.658 | 0.123 ± 0.002 | 2.878 ± 0.695 | 1.75 | |
| 15 | Cl | >30 | 3.257 ± 0.626 | 1.138 ± 0.004 | 1.53 | |||
| 16 | PhCH2NH | >30 | 0.718 ± 0.149 | 0.432 ± 0.025 | 4.76 | |||
| 17 | (CH3)2N | 2.903 ± 0.648 | 0.076 ± 0.017 | 1.146 ± 0.107 | 0.59 | |||
| 18 | CONHCH2Ph | >30 | >30 | >30 | 3.37 | |||
| 19 | CH2COOCH3 | 20.675 ± 1.865 | 13.460 ± 0.980 | 14.770 ± 3.010 | 2.55 | |||
| 20 | CH2CONHCH2Ph | >30 | >30 | 3.374 ± 205 | 1.87 | |||
| 21 | CH2CN | 8.657 ± 1.524 | 3.478 ± 0.312 | 5.957 ± 1.021 | 0.36 | |||
| 23 | (CH2)2Ph | 1.336 ± 0.279 | 1.174 ± 0.037 | 0.151 ± 0.015 | 0.66 | |||
| 24 | (CH2)3Ph | 0.692 ± 0.088 | 3.335 ± 0.749 | 1.275 ± 0.289 | 0.73 | |||
| 25 | (CH2)2Ph-4Cl | 10.812 ± 2.039 | 2.655 ± 0.381 | 2.106 ± 515 | 1.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinaci, A.; Buccioni, M.; Catarzi, D.; Cui, C.; Colotta, V.; Dal Ben, D.; Cescon, E.; Francucci, B.; Grieco, I.; Lambertucci, C.; et al. “Dual Anta-Inhibitors” of the A2A Adenosine Receptor and Casein Kinase CK1delta: Synthesis, Biological Evaluation, and Molecular Modeling Studies. Pharmaceuticals 2023, 16, 167. https://doi.org/10.3390/ph16020167
Spinaci A, Buccioni M, Catarzi D, Cui C, Colotta V, Dal Ben D, Cescon E, Francucci B, Grieco I, Lambertucci C, et al. “Dual Anta-Inhibitors” of the A2A Adenosine Receptor and Casein Kinase CK1delta: Synthesis, Biological Evaluation, and Molecular Modeling Studies. Pharmaceuticals. 2023; 16(2):167. https://doi.org/10.3390/ph16020167
Chicago/Turabian StyleSpinaci, Andrea, Michela Buccioni, Daniela Catarzi, Chang Cui, Vittoria Colotta, Diego Dal Ben, Eleonora Cescon, Beatrice Francucci, Ilenia Grieco, Catia Lambertucci, and et al. 2023. "“Dual Anta-Inhibitors” of the A2A Adenosine Receptor and Casein Kinase CK1delta: Synthesis, Biological Evaluation, and Molecular Modeling Studies" Pharmaceuticals 16, no. 2: 167. https://doi.org/10.3390/ph16020167