1. Introduction
Ephrin receptors (EPHR) represent the largest family of receptor tyrosine kinases (RTK) in mammals that regulate such diverse processes as cell migration, proliferation, survival and differentiation in multiple different cell types and tissues [
1,
2,
3]. Of the two classes of EPHR, there are nine EPHA receptors that bind five different Ephrin-A ligands and five EPHB receptors that bind three different Ephrin-B ligands [
4,
5]. EPHR are typical RTK that comprise an extracellular region with ligand binding and fibronectin homology domains, a short transmembrane spanning domain, and an intracellular region that contains a juxtamembrane (JM) regulatory sequence, a protein tyrosine kinase (PTK) domain and a C-terminal sterile alpha motif (SAM) domain (
Figure 1) [
1,
2,
3,
4,
5]. EPHR can activate multiple different intracellular signaling pathways that couple cell surface ligand recognition events to downstream cellular responses. These include activation of small GTP-binding proteins of the Rho and Ras families and activation of the phosphatidyl inositol 3-kinase (PI3K)-AKT signaling pathway [
1,
2,
6]. The activation or inhibition of these pathways, particularly Ras signaling, can vary depending on the cellular context [
1,
2,
6].
In the circulatory system, in vitro analyses, human genetic studies and mouse gene-targeting studies have each pointed to a pivotal function for EPHB4 in particular as a regulator of vascular development and function (see below). Moreover, in this role, there is now significant evidence that EPHB4 communicates with Ras p21 protein activator 1 (RASA1), also known as p120 Ras GTPase-activating protein (p120RasGAP), to inhibit Ras activation in endothelial cells (EC). Herein, we review this evidence and provide insights into possible molecular therapies for vascular disorders arising from impaired EPHB4-RASA1 signaling in humans.
2. EPHR-Mediated Inhibition of Ras Signaling In Vitro
RTK activate Ras through recruitment of Ras guanine nucleotide exchange factors (GEF) to cell membranes that switch membrane-tethered Ras from its inactive GDP-bound state to its active GTP-bound state [
7,
8]. In its GTP-bound state, Ras triggers the activation of downstream signaling pathways including the mitogen-activated protein kinase (MAPK) pathway that culminates in the phosphorylation and activation of extracellular signal-regulated serine/threonine protein kinases (ERKs) [
9]. Activated ERKs drive cellular responses through multiple mechanisms including phosphorylation and activation of nuclear transcription factors.
Several studies performed in cell lines in vitro have shown that EPHR can inhibit activation of the Ras-MAPK pathway triggered by distinct RTK [
10,
11,
12,
13,
14,
15,
16,
17]. This has been demonstrated for both EPHA and EPHB class receptors, including EPHB4, and in a variety of cell types including myoblasts, neurons, transformed cells and EC. In some studies, the ability of EPHR to inhibit Ras-MAPK activation was shown to be dependent upon expression of functional RASA1 (p120 RasGAP) [
10,
15,
16].
RASA1 is one member of family of ten different RasGAPs in mammals [
18]. It comprises two N-terminal Src homology-2 (SH2) domains separated by an SH3 domain, followed by pleckstrin homology (PH) and protein kinase C2 homology (C2) domains and a C-terminal catalytically active GAP domain (
Figure 1). As a RasGAP, RASA1 inactivates Ras by greatly augmenting its ability to hydrolyze bound GTP to GDP. Expression of a truncated form of RASA1 in neuronal cells blocked an ability of EPHB receptors to inhibit Ras-MAPK activation [
10]. In addition, knockdown of RASA1 in myoblasts prevented an ability of EPHA receptors to inhibit Ras-MAPK activation induced by insulin growth factor receptor 1 [
15]. For EPHB4, knockdown of RASA1 in the human umbilical vein EC (HUVEC) blocked an ability of EPHB4 to inhibit activation of the Ras-MAPK pathway [
16]. Interestingly, although EPHB4 inhibits Ras-MAPK signaling in EC and EC proliferation through a RASA1-depednedent mechanism, in other cell types such as hepatic stellate cells or MC7 breast cancer cells, EPHB4 activates the Ras-MAPK pathway resulting in increased proliferation [
16,
19]. In MC7 cells, EPHB4 couples to the Ras-MAPK pathway through protein phosphatase 2A (PP2A) because knockdown of PP2A blocks the EPHB4-induced Ras-MAPK response [
16]. An ability of EPHB4 to inhibit Ras-MAPK signaling in EC but activate the same pathway in MC7 cells cannot be explained on the grounds of differential expression of RASA1 and PP2A because both are well expressed in the respective cell types. Hence, the basis for this difference remains to be determined.
Notwithstanding the cell context-dependent signaling activity of EPHB4, the above studies are consistent with the notion of an EPHR–RASA1 inhibitory axis that regulates Ras-MAPK activation in different cell types. However, evidence for this mechanism as a regulator of Ras-MAPK activation in vivo is thus far limited to EPHB4 in EC.
3. Human Vascular Anomalies Caused by EPHB4 and RASA1 Mutations
Germline inactivating mutations of EPHB4 and RASA1 genes result in the same vascular anomalies in humans. This finding supports the idea that EPHB4 and RASA1 act together in the same pathway performing essentially the same function in the vasculature in vivo.
3.1. Capillary Malformation–Arteriovenous Malformation
Capillary malformation–arteriovenous malformation (CM–AVM) is an inherited autosomal dominant vascular anomaly with characteristic cutaneous CM and, in approximately one third of individuals, additional fast flow vascular lesions including AVMs and arteriovenous fistulas that can be life-threatening depending upon location [
20,
21,
22]. In rare cases, lymphatic vessel disorders are also present which include lymphatic malformation and disordered lymphatic vessel flow, chylous ascites and chylothorax (accumulation of lymph fluid in the peritoneal and pleural cavities, respectively) [
21,
23,
24,
25]. The first gene reported to be affected in CM–AVM was
RASA1 [
26]. The vast majority of
RASA1 mutations in CM–AVM are nonsense mutations, splice site substitutions and frame shift mutations that result in premature stop codons leading to nonsense-mediated RNA decay and predicted total loss of RASA1 protein directed by the mutated germline allele. A minority of mutations are missense mutations that are predicted to affect RASA1 function if not abundance (see below). To explain the autosomal dominant nature of inheritance as well as the sporadic nature of lesions in CM–AVM, a second hit model of CM–AVM pathogenesis was proposed in which inactivating somatic mutation of the inherited wild-type
RASA1 allele in EC during development is conceived to result in RASA1 null EC that drive lesion development [
20]. This possibility was first convincingly demonstrated in a study in which a nonsense
RASA1 mutation that was in trans to the inherited
RASA1 mutation was identified in EC derived from a micro-AVM in a patient with CM–AVM but was absent from EC from non-lesional tissue in the same patient [
27].
RASA1 gene mutations account for approximately 70 percent of CM–AVM cases. More recently,
EPHB4 gene mutations have been identified to be responsible for the remaining 30 percent of CM–AVM cases [
28]. Approximately half of the
EPHB4 mutations in CM–AVM are predicted to result in loss of protein expression consequent to nonsense-mediated RNA decay, whereas the remaining
EPHB4 mutations are missense mutations. CM–AVM cases caused by
RASA1 or
EPHB4 mutations have been named CM–AVM1 and CM–AVM2 respectively to distinguish the two genetic causes. However, CM–AVM1 and CM–AVM2 are phenotypically indistinguishable except for the occurrence of blood vascular telangiectasias in some CM–AVM2 patients.
3.2. Other Vascular Anomalies
The vein of Galen malformation (VGAM) is the most common AVM in pediatric patients [
29,
30]. Although VGAM has been described in CM–AVM, other cases may present without the CM that are pathognomonic for CM–AVM. Inactivating germline mutations of
EPHB4 and
RASA1 have both been reported in VGAM [
21,
31,
32,
33,
34]. In addition, inactivating mutations of
EPHB4 and
RASA1 have both been reported in patients with lymphatic vessel disorders without CM–AVM. For
EPHB4, these include lymphatic-related hydrops fetalis (LRHF, accumulation of fluid in the fetus because of lymphatic dysfunction), central conducting lymphatic anomaly (a lymphatic vessel flow disorder) and lymphedema (tissue edema consequent to lymphatic dysfunction) [
35,
36,
37,
38]. Similarly, germline inactivating
RASA1 mutations have been reported in LRHF and lymphedema [
39,
40].
4. Mouse Genetic Studies
Mouse genetic studies provide further evidence for an inhibitory EPHB4–RASA1 axis in the vasculature in vivo. With few exceptions, genetic disruption of EPHB4 or RASA1 results in similar if not identical phenotypes. Each of these is described below.
4.1. Developmental Angiogenesis
Constitutive EPHB4-deficient and RASA1-deficient mice both die at embryonic day 10.5 (E10.5) of development from cardiovascular defects. The salient phenotype in both models is blocked developmental angiogenesis in which primitive vascular plexuses that are generated through the process of vasculogenesis fail to become remodeled into hierarchical arterial-capillary-venous networks [
41,
42]. The same phenotype was observed in mice constitutively deficient in the EPHB4 ligand, ephrin B2 [
41,
43,
44]. In addition, the same phenotype was noted in R780Q RASA1 knockin mice that express a form of RASA1 that is unable to promote Ras hydrolysis of GTP, but which would theoretically retain all other putative Ras-independent functions [
45]. This latter finding shows that impaired developmental angiogenesis in constitutive RASA1-deficient mice results from an inability of RASA1 to promote Ras inactivation specifically. By extension, vascular anomalies in CM–AVM1 are also likely to be consequent to an inability of RASA1 to inactivate Ras and not to loss of a Ras-independent function of RASA1, e.g., Rho family GTPase regulation as suggested by others [
46].
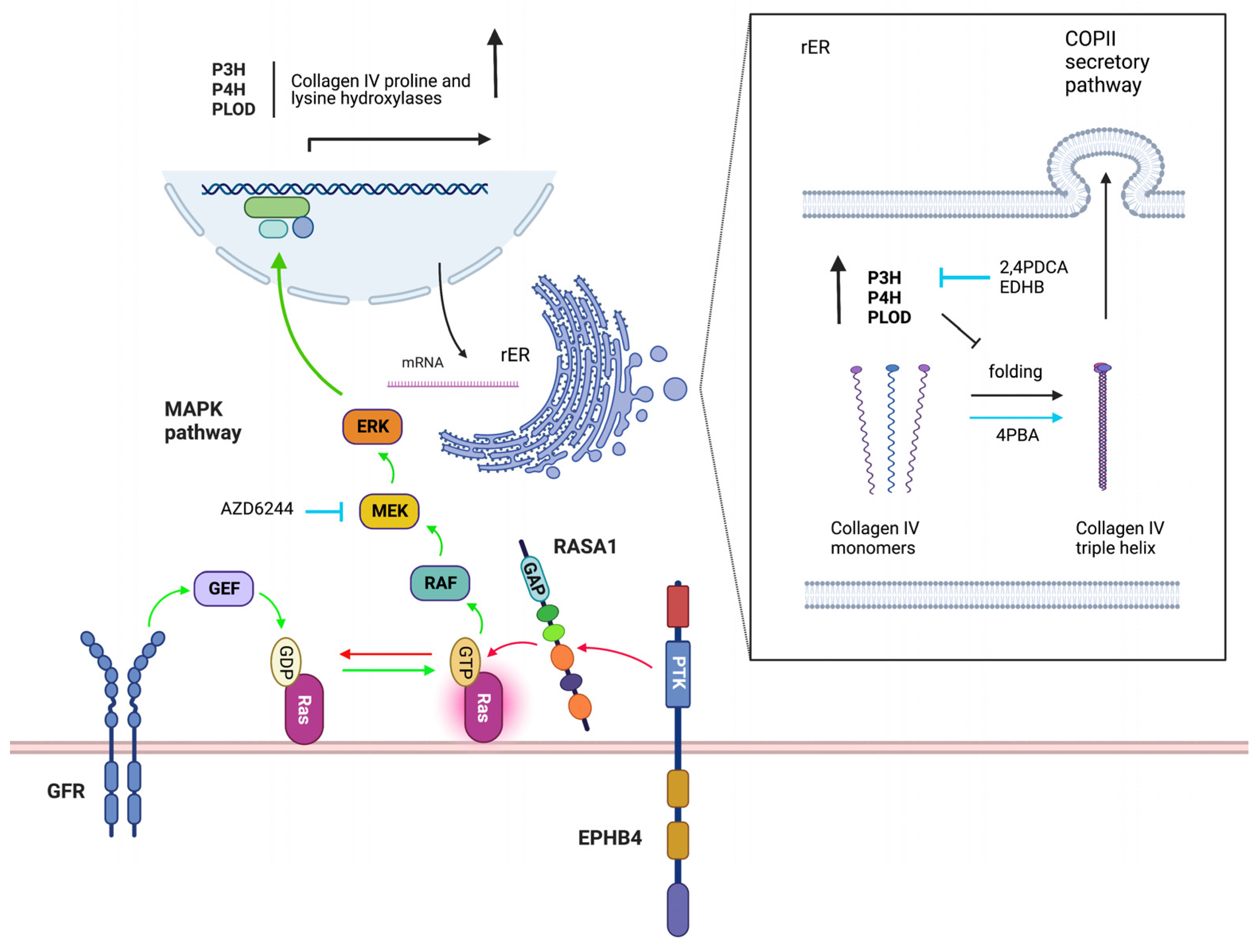
To study the mechanism by which loss of EPHB4 and RASA1 impairs developmental angiogenesis, Chen et al. used conditional EPHB4- or RASA1-deficient mouse models to disrupt the respective genes within EC at E13.5 when vasculogenesis has ceased and developmental angiogenesis predominates [
47,
48]. In both models, by E18.5, extensive cutaneous hemorrhage was observed that was associated with apoptosis of blood vascular EC (BEC) and loss of lymphatic EC (LEC). In both cases, EC failed to export the vascular basement membrane protein, collagen IV, that instead was trapped within the EC endoplasmic reticulum (ER). The accumulated collagen triggered EC apoptosis though induction of an unresolved unfolded protein response. In addition, the much-reduced amounts of collagen IV in vascular basement membranes resulted in detachment of EC and apoptosis by anoikis. Anoikis (Greek for “without a home”) is the default form of apoptosis that results when a normally adherent cell fails to attach to a substratum [
49]. The small molecular weight chaperone, 4 phenyl butyric acid (4PBA), that had been shown to promote folding and export of ER-trapped misfolded point mutant forms collagen IV beforehand, rescued collagen IV export and EC apoptosis in induced EPHB4- and RASA1-deficient embryos [
47,
48,
50,
51]. This finding demonstrated that collagen IV retention in the ER was likely a result of collagen IV misfolding (
Figure 2).
Consistent with the model of EPHB4 as a negative regulator of Ras-MAPK signaling that acts through communication with RASA1, loss of EPHB4 or RASA1 both resulted in greatly augmented activation of ERK MAPK in EC during developmental angiogenesis [
47,
48]. Moreover, drugs that inhibit MAPK activation also allowed normal collagen IV export from EC and rescued EC apoptosis showing that dysregulated Ras-MAK activation was the key initiating event in the development of vascular phenotypes in induced EPHB4- and RASA1-deficient embryos [
47,
48] (
Figure 2).
To understand how dysregulated Ras-MAPK activation might lead to collagen IV misfolding, proteomic analyses were performed upon RASA1-deficient and control cutaneous EC during developmental angiogenesis [
48]. Intriguingly, of seven members of a family of nine collagen proline and lysine hydroxylases that could be detected, all were increased in abundance in RASA1-deficient compared to control EC. Hydroxylation of collagens on prolines and lysine residues in the ER promotes folding of collagen monomers into trimeric promoters and their export from the cell via the COPII secretion pathway [
52,
53,
54]. However, excessive hydroxylation may impair collagen folding and cellular export. In agreement with a model in which increased abundance of collagen proline and lysine hydroxylases in the EC ER results in over-hydroxylation of collagen IV monomers that blocks collagen IV export, drugs that inhibit all members of the collagen proline and lysine hydroxylase family (part of the larger group of 2 oxoglutarate-dependent oxygenases) also rescued collagen IV export and EC apoptosis during developmental angiogenesis in EPHB4- and RASA1-deficient embryos [
48,
55] (
Figure 2).
4.2. Development and Maintenance of Vascular Valves
The collection of lymphatic vessels and veins that are invested with intraluminal semilunar valves can facilitate directional fluid flow toward the blood vasculature and heart, respectively [
56,
57]. In addition, specialized valves, known as lymphovenous valves, prevent backflow of venous blood into lymphatic vessels at their point of connection with the venous vasculature in the region of intersection of jugular veins, the subclavian vein and the superior vena cava [
57,
58,
59]. Studies of conditional EPHB4- and RASA1-deficient mice have revealed that EPHB4 and RASA1 are both required for the development all three types of valve [
36,
60,
61,
62,
63]. Furthermore, all three types of valve fail to develop properly in embryos induced to express R780Q RASA1 alone in the vasculature [
60,
62]. Consistent with this, blocked development of lymphovenous valves and venous valves in induced RASA1-deficient embryos can be rescued by inhibitors of Ras-MAPK signaling [
60]. In addition, drugs that inhibit the activity of collagen IV-modifying proline and lysine hydroxylases, and 4PBA can rescue development of lymphovenous valves and venous valves in induced RASA1-deficient embryos [
60]. Therefore, a similar mechanism invoked to explain impaired developmental angiogenesis likely also accounts for impaired development of lymphovenous and venous valves. In this regard, an inability of valve leaflet-forming EC to deposit collagen IV into the extracellular matrix core of the developing valve leaflet would be expected to lead to apoptotic death of leaflet cells and block valve leaflet elongation.
As well as development, maintenance of lymphatic vessel valves in adult mice also depends upon EPHB4 and RASA1 and catalytically active RASA1 specifically [
62] (and unpublished). In induced RASA1-deficient adult mice, it is estimated that popliteal lymphatic vessel valve leaflets lose an average of one LEC per week resulting in progressive valve shortening until a threshold is reached at which point valves are unable to prevent fluid backflow [
62]. Consequently, mice exhibit grossly disrupted lymphatic flow patterns and eventually die from chylothorax [
64]. Similarly, induced EPHB4-deficient mice show abnormal lymphatic vessel flow and at least some mice succumb to chylothorax (unpublished).
4.3. Neonatal and Pathological Angiogenesis
The disruption of either
Eph4 or
Rasa1 genes in mice or the switch to expression of R780Q RASA1 alone in the neonatal period in mice results in impaired retinal angiogenesis as evidenced by reduced EC coverage and fewer vessel branch points [
47,
48]. Retinas from both types of mice show increased numbers of “empty sleeves” that consist of thin sheaths of collagen IV without EC. These findings are consistent with a function for EPHB4 and RASA1 in export of collagen IV from EC during active angiogenesis. In this regard, an inability of EC to export collagen IV during retinal angiogenesis would result in loss of EC through detachment and apoptotic death resulting in the “empty sleeve” appearance.
EPHB4 and RASA1 are also required for pathological angiogenesis necessary for the growth of solid tumors in adult mice [
47,
48]. In the absence of EPHB4 or RASA1, tumor angiogenesis is impaired and is associated with retention of collagen IV in BEC. Significantly, 4PBA restores collagen IV export, tumor angiogenesis and tumor growth [
47,
48]. These findings represent an additional example of a requirement for EPHB4 and RASA1 for active angiogenesis and provide further evidence that they function in the same molecular pathway in vivo.
4.4. Differences between EPHB4- and RASA1-Deficient Mouse Models
Some differences in phenotype between EPHB4-deficient and RASA1-deficient mouse models are apparent. Notably, induced EC-specific disruption of EPHB4 in adult mice results in rupturing and remodeling of cardiac capillaries and cardiac hypertrophy [
17]. In contrast, cardiac hypertrophy has not been observed in induced EC-specific RASA1-deficient mice [
64]. In addition, it has been reported that induced EC-specific disruption of EPHB4 in postnatal mice leads to loss of junctional integrity between LEC in collecting lymphatic vessels [
61]. In contrast, no such loss of LEC junctional integrity has been observed in induced EC-specific RASA1-deficient mice [
62,
64]. Potentially, such differences could be explained by loss of EPHB4-mediated activation of distinct signaling pathways, such as the PI3K signaling pathway, in EC [
17,
65]. Alternatively, or in addition, some differences may relate to a loss of reverse signaling through Ephrin B2 in EPHB4-deficient mice [
2,
66,
67].
5. Mechanism of EPHB4–RASA1 Communication in the Vasculature
EPHB4 and RASA1 are known to interact physically during EPHB4 signal transduction [
11,
47]. The interaction is mediated by RASA1 SH2 domain recognition of a pair of autophosphorylated tyrosine residues in the EPHB4 JM region (
Figure 3). Therefore, one obvious model that could account for the functional relationship between EPHB4 and RASA1 in the vasculature is that EPHB4 serves to recruit RASA1 to the plasma membrane whereupon it would be juxtaposed to Ras-GTP that would facilitate Ras inactivation. To test this model, Chen et al. generated a knockin
Ephb4 mutant mouse allele that encodes a form of EPHB4 that is unable to bind RASA1 [
47]. Because phosphorylation of the same EPHB4 JM tyrosine residues is necessary to switch EPHB4 from an inactive to an active open conformation, simple mutation of these tyrosines to phenylalanine would not be informative, as such a receptor would be devoid of kinase activity, which would complicate interpretation of results [
47,
68,
69,
70]. Therefore, in addition to tyrosine to phenylalanine substitutions, two key proline residues in the JM region were substituted for glycine in the mutant receptor [
68,
69,
70]. These additional mutations circumvented a requirement for phosphorylation of JM tyrosine residues for EPHB4 kinase activity [
47].
Surprisingly, mice that were homozygous for the RASA1-binding deficient EPHB4 receptor showed normal development of the blood vascular system. In addition, mice exhibited normal retinal angiogenesis and pathological angiogenesis toward solid tumors [
47]. Therefore, these results demonstrate that whatever the basis for the functional relationship between EPHB4 and RASA1 in the blood vasculature, it is not require physical interaction between the two molecules. Considering alternative possibilities, it is of note that of the small number of
RASA1 missense mutations that have been reported in CM–AVM, most are concentrated within the PH and C2 domains of the protein that are implicated in membrane lipid binding [
20,
26,
34,
71]. An alternative model, therefore, is that RASA1 is targeted to Ras-GTP though recognition of membrane lipids generated during course of EPHB4 signaling (
Figure 3). However, experimental evidence for this model awaits.
6. Pathogenesis of Vascular Anomalies in Patients with EPHB4 and RASA1 Mutations
How second hit mutation of
EPHB4 or
RASA1 genes in patients with inherited mutated
EPHB4 or
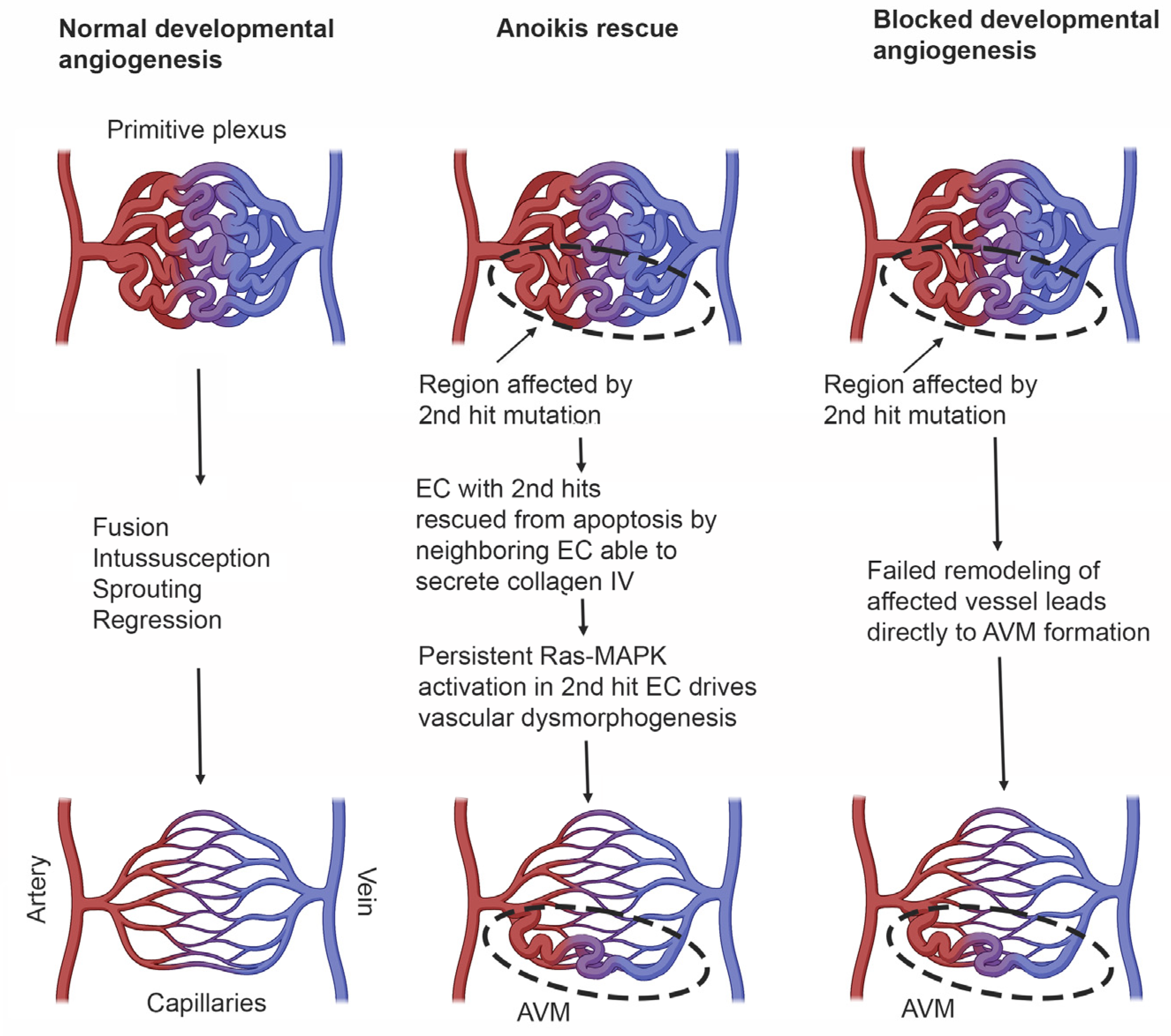
RASA1 genes, respectively, gives rise to vascular lesions is uncertain. In most cases, it seems reasonable to propose that the exact nature of lesions, i.e., CM, AVM or lymphatic abnormalities or combinations thereof, depends upon the location of the second hit or hits, i.e., which vascular bed, as well as the time during development that the second hit(s) mutation occurs. However, reconciling how in humans second hit mutation in EC during developmental angiogenesis leads to vascular malformation, whereas in mice loss of EC EPHB4 or RASA1 during developmental angiogenesis results in apoptotic death of EC, is more challenging. One possible explanation for this apparent difference is that in humans, EC that have acquired a second hit mutation are rescued from anoikis on the basis that adjacent EC that have not acquired a second hit mutation are able to export collagen IV normally for deposition in the basement membrane. On the other hand, apoptotic death of EC resulting from an unfolded protein response would not be subverted by an ability of adjacent EC without second hit mutations to continue to engage in collagen IV export. In the case that EC death is substantively rescued through avoidance of anoikis, Ras-MAPK signaling must drive vascular malformation through some undetermined mechanism unrelated to export of collagen IV (
Figure 4).
An alternative explanation for the origin of AVMs relates to a potential inability to remodel individual vessels in primitive vascular plexuses. In this regard, should a second hit mutation occur early enough in blood or lymphatic vasculogenesis, it is possible that the majority if not all EC within an individual vessel of a primitive plexus may not express any functional EPHB4 or RASA1. In such a circumstance, remodeling of that vessel by sprouting angiogenesis would not be possible because the demand for de novo synthesis of collagen IV by EC engaging in this event would lead to their apoptotic death (
Figure 4). The result would be direct connection between arteries and veins without an intervening capillary bed. For the lymphatic vasculature, it is also possible that, if second hit mutations were to occur early enough, all putative valve-forming cells within a zone of a collecting lymphatic vessel where valve formation would be required would be EPHB4 or RASA1 null. As such, valvulogenesis would not proceed as this would also necessitate collagen IV synthesis. In this way, second hit mutation could contribute to some lymphatic pathologies.
Last, it is of note that in some families with
EPHB4 mutations, affected individuals in different generations present only with lymphatic abnormalities, i.e., LRHF or lymphedema [
36,
37,
38]. This finding is difficult to explain the context of the second hit model of disease expression because it demands that the second hit consistently occurs in lymphatic but not blood vascular beds in each affected individual in these families. Nor is there any evidence that mutant forms of EPHB4 in these families show dominant negative behavior that might otherwise provide an alternative explanation for the lymphatic only phenotype. Hence, the origin of lymphatic abnormalities in these patients remains to be determined.
7. Prospects for Drug Therapy of Vascular Anomalies Caused by EPHB4 or RASA1 Mutation
Based on studies performed in mice, drugs that dampen the activation of MAPK or promote the folding of collagen IV in the EC ER, either through blockade of collagen IV proline or lysine hydroxylases or through direct action on collagen IV itself, have potential for the prevention and treatment of vascular anomalies in patients with inherited EPHB4 and RASA1 gene mutations. However, the utility of each will depend upon which of the models of vascular pathogenesis outlined above is likely to apply. In the case that EC with second hit mutations can be rescued from anoikis by proximal EC without second mutations, drugs that promote collagen IV folding are not likely to be of benefit. However, in the alternative hypothesis of impaired remodeling of primitive vascular plexuses, these drugs and MAPK pathway inhibitors could be useful.
Another consideration is the time at which drugs would need to be administered for preventative or therapeutic effect. In most cases, vascular lesions in patients arise during development and are present at birth, although can be exacerbated during postnatal life by events such as puberty or pregnancy. Therefore, prevention of lesion development would require drug administration during fetal development. The extent to which maintenance or growth of lesions requires ongoing dysregulated signal transduction through the Ras-MAPK pathway is unknown. Thus, it is unclear if drug intervention would be effective for the treatment of established vascular lesions. However, this is also likely to depend upon which model of pathogenesis applies. In the case of anoikis rescue, MAPK pathway inhibitors may still have utility. Understanding which model of pathogenesis applies awaits the development of additional mouse models that recapitulate the second hit that is considered to account for most vascular anomalies arising from EPHB4 or RASA1 mutation.
8. Summary
A preponderance of evidence from in vitro and in vivo studies now supports the view that the principal function of EPHB4 in the vasculature is to negatively regulate the activation of the Ras-MAPK pathway in EC and that in this role RASA1 is the EPHB4 effector. Loss of EPHB4 or RASA1 function in EC results in dysregulated Ras-MAPK signaling that drives the development of vascular abnormalities in CM–AVM and other vascular disorders. In mice, the dysregulated Ras-MAPK signaling inhibits EC export of collagen IV, which at least in part is responsible for the development of vascular abnormalities. Whether or not the same mechanism is responsible for the development of vascular abnormalities in humans remain to be established. Conceivably, blocked EC export of collagen IV during developmental angiogenesis could prevent remodeling of primitive vascular plexuses into hierarchical arterial-capillary-venous networks, which could account for some types of vascular abnormalities resulting from loss of RASA1 or EPHB4 function in humans. Alternatively, because loss of RASA1 or EPHB4 function is initially expected to be localized to individual EC in human vascular disorders, it is possible that adjacent RASA1- or EPHB4-sufficient EC can rescue RASA1- or EPHB4- null EC from apoptosis through provision of collagen IV in trans. In this latter situation, dysregulated Ras-MAPK signaling may drive vascular abnormalities through yet to be characterized mechanisms. Prospectively, drugs that inhibit the Ras-MAPK pathway or promote EC export of collagen IV could be of benefit for the prevention and treatment of patients with vascular anomalies resulting from inherited and somatic mutations in EPHB4 or RASA1 genes.
Author Contributions
Writing—original draft preparation, P.D.K.; review and editing, D.C., M.A.V.d.E. and N.L.L.; funding acquisition, P.D.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health, grant numbers HL146352 and HL120888.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable in this paper.
Acknowledgments
All figures were created with BioRender.com (accessed on 22 January 2023).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kania, A.; Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barquilla, A.; Pasquale, E.B. Eph receptors and ephrins: Therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.; Huang, Q.; Nussinov, R.; Ma, B. Promiscuous and specific recognition among ephrins and Eph receptors. Biochim. Biophys. Acta. 2014, 1844, 1729–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohani, N.; Parmeggiani, A.; Winklbauer, R.; Fagotto, F. Variable combinations of specific ephrin ligand/Eph receptor pairs control embryonic tissue separation. PLoS Biol. 2014, 12, e1001955. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene. 2019, 38, 6567–6584. [Google Scholar] [CrossRef] [Green Version]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Elowe, S.; Holland, S.J.; Kulkarni, S.; Pawson, T. Downregulation of the Ras-mitogen-activated protein kinase pathway by the EphB2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Mol. Cell Biol. 2001, 21, 7429–7441. [Google Scholar] [CrossRef]
- Kim, I.; Ryu, Y.S.; Kwak, H.J.; Ahn, S.Y.; Oh, J.L.; Yancopoulos, G.D.; Gale, N.W.; Koh, G.Y. EphB ligand, ephrinB2, suppresses the VEGF- and angiopoietin 1-induced Ras/mitogen-activated protein kinase pathway in venous endothelial cells. FASEB J. 2002, 16, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Macrae, M.; Neve, R.M.; Rodriguez-Viciana, P.; Haqq, C.; Yeh, J.; Chen, C.; Gray, J.W.; McCormick, F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005, 8, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, C.; Anastasiadou, S.; Knoll, B. Ephrin-A5 suppresses neurotrophin evoked neuronal motility, ERK activation and gene expression. PLoS ONE 2011, 6, e26089. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Wei, B.R.; Peehl, D.M.; Li, Q.; Alexandrou, T.; Schelling, J.R.; Burnett, E.; Wang, B. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat. Cell Biol. 2001, 3, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Koyama, T.; Wakayama, Y.; Fukuhara, S.; Mochizuki, N. EphrinA/EphA signal facilitates insulin-like growth factor-I-induced myogenic differentiation through suppression of the Ras/extracellular signal-regulated kinase 1/2 cascade in myoblast cell lines. Mol. Biol. Cell. 2011, 22, 3508–3519. [Google Scholar] [CrossRef]
- Xiao, Z.; Carrasco, R.; Kinneer, K.; Sabol, D.; Jallal, B.; Coats, S.; Tice, D.A. EphB4 promotes or suppresses Ras/MEK/ERK pathway in a context-dependent manner: Implications for EphB4 as a cancer target. Cancer Biol. Ther. 2012, 13, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Luxan, G.; Stewen, J.; Diaz, N.; Kato, K.; Maney, S.K.; Aravamudhan, A.; Berkenfeld, F.; Nagelmann, N.; Drexler, H.C.; Zeuschner, D.; et al. Endothelial EphB4 maintains vascular integrity and transport function in adult heart. Elife 2019, 8, e45863. [Google Scholar] [CrossRef]
- King, P.D.; Lubeck, B.A.; Lapinski, P.E. Nonredundant functions for Ras GTPase-activating proteins in tissue homeostasis. Sci Signal 2013, 6, re1. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Shergill, U.; Thakur, L.; Sinha, S.; Urrutia, R.; Mukhopadhyay, D.; Shah, V.H. Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G908–G915. [Google Scholar] [CrossRef] [Green Version]
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.J.; Irvine, A.D.; Baselga, E.; et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013, 34, 1632–1641. [Google Scholar] [CrossRef]
- Revencu, N.; Boon, L.M.; Mulliken, J.B.; Enjolras, O.; Cordisco, M.R.; Burrows, P.E.; Clapuyt, P.; Hammer, F.; Dubois, J.; Baselga, E.; et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum. Mutat. 2008, 29, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooderchak-Donahue, W.L.; Johnson, P.; McDonald, J.; Blei, F.; Berenstein, A.; Sorscher, M.; Mayer, J.; Scheuerle, A.E.; Lewis, T.; Grimmer, J.F.; et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur. J. Hum. Genet. 2018, 26, 1521–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, P.E.; Gonzalez-Garay, M.L.; Rasmussen, J.C.; Aldrich, M.B.; Guilliod, R.; Maus, E.A.; Fife, C.E.; Kwon, S.; Lapinski, P.E.; King, P.D.; et al. Lymphatic abnormalities are associated with RASA1 gene mutations in mouse and man. Proc. Natl. Acad. Sci. USA 2013, 110, 8621–8626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wijn, R.S.; Oduber, C.E.; Breugem, C.C.; Alders, M.; Hennekam, R.C.; van der Horst, C.M. Phenotypic variability in a family with capillary malformations caused by a mutation in the RASA1 gene. Eur. J. Med. Genet. 2012, 55, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Macmurdo, C.F.; Wooderchak-Donahue, W.; Bayrak-Toydemir, P.; Le, J.; Wallenstein, M.B.; Milla, C.; Teng, J.M.C.; Bernstein, J.A.; Stevenson, D.A. RASA1 somatic mutation and variable expressivity in capillary malformation/arteriovenous malformation (CM/AVM) syndrome. Am. J. Med. Gent. A. 2016, 170, 1450–1454. [Google Scholar] [CrossRef]
- Eerola, I.; Boon, L.M.; Mulliken, J.B.; Burrows, P.E.; Dompmartin, A.; Watanabe, S.; Vanwijck, R.; Vikkula, M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 2003, 73, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, P.E.; Doosti, A.; Salato, V.; North, P.; Burrows, P.E.; King, P.D. Somatic second hit mutation of RASA1 in vascular endothelial cells in capillary malformation-arteriovenous malformation. Eur. J. Med. Genet. 2018, 61, 11–16. [Google Scholar] [CrossRef]
- Amyere, M.; Revencu, N.; Helaers, R.; Pairet, E.; Baselga, E.; Cordisco, M.; Chung, W.; Dubois, J.; Lacour, J.-P.; Martorell, L.; et al. Germline Loss-of-Function Mutations in EPHB4 Cause a Second Form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) Deregulating RAS-MAPK Signaling. Circulation 2017, 136, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Deloison, B.; Chalouhi, G.E.; Sonigo, P.; Zerah, M.; Millischer, A.E.; Dumez, Y.; Brunelle, F.; Ville, Y. Hidden mortality of prenatally diagnosed vein of Galen aneurysmal malformation: Retrospective study and review of the literature. Ultrasound Obstet. Gynecol. 2012, 40, 652–658. [Google Scholar] [CrossRef]
- Long, D.M.; Seljeskog, E.L.; Chou, S.N.; French, L.A. Giant arteriovenous malformations of infancy and childhood. J. Neurosurg. 1974, 40, 304–312. [Google Scholar] [CrossRef]
- Duran, D.; Zeng, X.; Jin, S.C.; Choi, J.; Nelson-Williams, C.; Yatsula, B.; Gaillard, J.; Furey, C.G.; Lu, Q.; Timberlake, A.T.; et al. Mutations in Chromatin Modifier and Ephrin Signaling Genes in Vein of Galen Malformation. Neuron 2019, 101, 429–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanti, A.; Ozanne, A.; Grondin, C.; Saliou, G.; Quevarec, L.; Maurey, H.; Aubourg, P.; Benachi, A.; Gut, M.; Gut, I.; et al. Loss of function mutations in EPHB4 are responsible for vein of Galen aneurysmal malformation. Brain 2018, 141, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Tas, B.; Starnoni, D.; Smajda, S.; Vivanti, A.J.; Adamsbaum, C.; Eyries, M.; Melki, J.; Tawk, M.; Ozanne, A.; Revencu, N.; et al. Arteriovenous Cerebral High Flow Shunts in Children: From Genotype to Phenotype. Front. Pediatr. 2022, 10, 871565. [Google Scholar] [CrossRef] [PubMed]
- Kwong, Y.; Cartmill, M.; Jaspan, T.; Suri, M. Fetal MRI demonstrating vein of Galen malformations in two successive pregnancies--a previously unreported occurrence. Childs Nerv. Syst. 2015, 31, 1033–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wenger, T.L.; Seiler, C.; March, M.E.; Gutierrez-Uzquiza, A.; Kao, C.; Bhoj, E.; Tian, L.; Rosenbach, M.; Liu, Y.; et al. Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly. Hum. Mol. Genet. 2018, 27, 3233–3245. [Google Scholar] [CrossRef]
- Martin-Almedina, S.; Martinez-Corral, I.; Holdhus, R.; Vicente, A.; Fotiou, E.; Lin, S.; Petersen, K.; Simpson, M.A.; Hoischen, A.; Gilissen, C.; et al. EPHB4 kinase-inactivating mutations cause autosomal dominant lymphatic-related hydrops fetalis. J. Clin. Investig. 2016, 126, 3080–3088. [Google Scholar] [CrossRef]
- Martin-Almedina, S.; Ogmen, K.; Sackey, E.; Grigoriadis, D.; Karapouliou, C.; Nadarajah, N.; Ebbing, C.; Lord, J.; Mellis, R.; Kortuem, F.; et al. Janus-faced EPHB4-associated disorders: Novel pathogenic variants and unreported intrafamilial overlapping phenotypes. Genet. Med. 2021, 23, 1315–1324. [Google Scholar] [CrossRef]
- Greene, A.K.; Brouillard, P.; Sudduth, C.L.; Smits, P.J.; Konczyk, D.J.; Vikkula, M. EPHB4 mutation causes adult and adolescent-onset primary lymphedema. Am. J. Med. Genet. A. 2021, 185, 3810–3813. [Google Scholar] [CrossRef]
- Gallipoli, A.; MacLean, G.; Walia, J.S.; Sehgal, A. Congenital Chylothorax and Hydrops Fetalis: A Novel Neonatal Presentation of RASA1 Mutation. Pediatrics 2021, 147, 3. [Google Scholar] [CrossRef]
- Westphal, D.S.; Bergmann, K.; Martens, E.; Ibrahim, T. A case report of RASA1-associated inherited lymphoedema with recurrent life-threatening lymphangitis. Eur. Heart J. Case Rep. 2021, 5, ytab451. [Google Scholar] [CrossRef]
- Gerety, S.S.; Wang, H.U.; Chen, Z.F.; Anderson, D.J. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999, 4, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Rossi, D.J.; Holmyard, D.P.; Puri, M.C.; Mbamalu, G.; Harpal, K.; Shih, T.S.; Jacks, T.; Pawson, T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995, 377, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, B.A.; Lapinski, P.E.; Bauler, T.J.; Oliver, J.A.; Hughes, E.D.; Saunders, T.L.; King, P.D. Blood vascular abnormalities in Rasa1(R780Q) knockin mice: Implications for the pathogenesis of capillary malformation-arteriovenous malformation. Am. J. Pathol. 2014, 184, 3163–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisa-Beygi, S.; Vo, N.J.; Link, B.A. RhoA activation-mediated vascular permeability in capillary malformation-arteriovenous malformation syndrome: A hypothesis. Drug. Discov. Today. 2021, 26, 1790–1793. [Google Scholar] [CrossRef]
- Chen, D.; Hughes, E.D.; Saunders, T.L.; Wu, J.; Vasquez, M.N.H.; Makinen, T.; King, P.D. Angiogenesis depends upon EPHB4-mediated export of collagen IV from vascular endothelial cells. JCI Insight. 2022, 7, 4. [Google Scholar] [CrossRef]
- Chen, D.; Teng, J.M.; North, P.E.; Lapinski, P.E.; King, P.D. RASA1-dependent cellular export of collagen IV controls blood and lymphatic vascular development. J. Clin. Invest. 2019, 130, 3545–3561. [Google Scholar] [CrossRef] [Green Version]
- Michel, J.B. Anoikis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef] [Green Version]
- Jeanne, M.; Jorgensen, J.; Gould, D.B. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation 2015, 131, 1555–1565. [Google Scholar] [CrossRef]
- Kuo, D.S.; Labelle-Dumais, C.; Mao, M.; Jeanne, M.; Kauffman, W.B.; Allen, J.; Favor, J.; Gould, D.B. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum. Mol. Genet. 2014, 23, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Chioran, A.; Duncan, S.; Catalano, A.; Brown, T.J.; Ringuette, M.J. Collagen IV trafficking: The inside-out and beyond story. Dev. Biol. 2017, 431, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Bachinger, H.P. A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta. 2013, 1833, 2479–2491. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef]
- Bazigou, E.; Makinen, T. Flow control in our vessels: Vascular valves make sure there is no way back. Cell Mol. Life Sci. 2013, 70, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Geng, X.; Cha, B.; Mahamud, M.R.; Srinivasan, R.S. Intraluminal valves: Development, function and disease. Dis. Model Mech. 2017, 10, 1273–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, X.; Cha, B.; Mahamud, M.R.; Lim, K.C.; Silasi-Mansat, R.; Uddin, M.K.M.; Miura, N.; Xia, L.; Simon, A.M.; Engel, J.D.; et al. Multiple mouse models of primary lymphedema exhibit distinct defects in lymphovenous valve development. Dev. Biol. 2016, 409, 218–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, R.S.; Oliver, G. Prox1 dosage controls the number of lymphatic endothelial cell progenitors and the formation of the lymphovenous valves. Genes Dev. 2011, 25, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Geng, X.; Lapinski, P.E.; Davis, M.J.; Srinivasan, R.S.; King, P.D. RASA1-driven cellular export of collagen IV is required for the development of lymphovenous and venous valves in mice. Development 2020, 147, 23. [Google Scholar] [CrossRef]
- Frye, M.; Stritt, S.; Ortsater, H.; Hernandez Vasquez, M.; Kaakinen, M.; Vicente, A.; Wiseman, J.; Eklund, L.; Vestweber, D.; Martínez-Torrecuadrada, J.L.; et al. EphrinB2-EphB4 signalling provides Rho-mediated homeostatic control of lymphatic endothelial cell junction integrity. Elife 2020, 9, e57732. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, P.E.; Lubeck, B.A.; Chen, D.; Doosti, A.; Zawieja, S.D.; Davis, M.J.; King, P.D. RASA1 regulates the function of lymphatic vessel valves in mice. J. Clin. Invest. 2017, 127, 2569–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, O.; Walker, J.; Seet, C.; Ikram, M.; Kuchta, A.; Arnold, A.; Hernández-Vásquez, M.; Frye, M.; Vizcay-Barrena, G.; Fleck, R.A.; et al. Mutations in EPHB4 cause human venous valve aplasia. JCI Insight. 2021, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, P.E.; Kwon, S.; Lubeck, B.A.; Wilkinson, J.E.; Srinivasan, R.S.; Sevick-Muraca, E.; King, P.D. RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J. Clin. Invest. 2012, 122, 733–747. [Google Scholar] [CrossRef]
- Steinle, J.J.; Meininger, C.J.; Forough, R.; Wu, G.; Wu, M.H.; Granger, H.J. Eph B4 receptor signaling mediates endothelial cell migration and proliferation via the phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 2002, 277, 43830–43835. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef]
- Foo, S.S.; Turner, C.J.; Adams, S.; Compagni, A.; Aubyn, D.; Kogata, N.; Lindblom, P.; Shani, M.; Zicha, D.; Adams, R.H. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 2006, 124, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Binns, K.L.; Taylor, P.P.; Sicheri, F.; Pawson, T.; Holland, S.J. Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors. Mol. Cell. Biol. 2000, 20, 4791–4805. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, S.; Wybenga-Groot, L.E.; Warner, N.; Lin, H.; Pawson, T.; Forman-Kay, J.D.; Sicheri, F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006, 25, 4686–4696. [Google Scholar] [CrossRef] [Green Version]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.; Pawson, T.; Sicheri, F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 2001, 106, 745–757. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.; Stevenson, D.A.; McDonald, J.; Grimmer, J.F.; Gedge, F.; Bayrak-Toydemir, P. RASA1 analysis: Clinical and molecular findings in a series of consecutive cases. Eur. J. Med. Genet. 2012, 55, 91–95. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


{kind=link}
{kind=link}
{kind=link}
{kind=link}



