
Cooccurring Type 1 Diabetes Mellitus and Autoimmune Thyroiditis in a Girl with Craniofrontonasal Syndrome: Are EFNB1 Variants Associated with Autoimmunity?

 , ,
, , 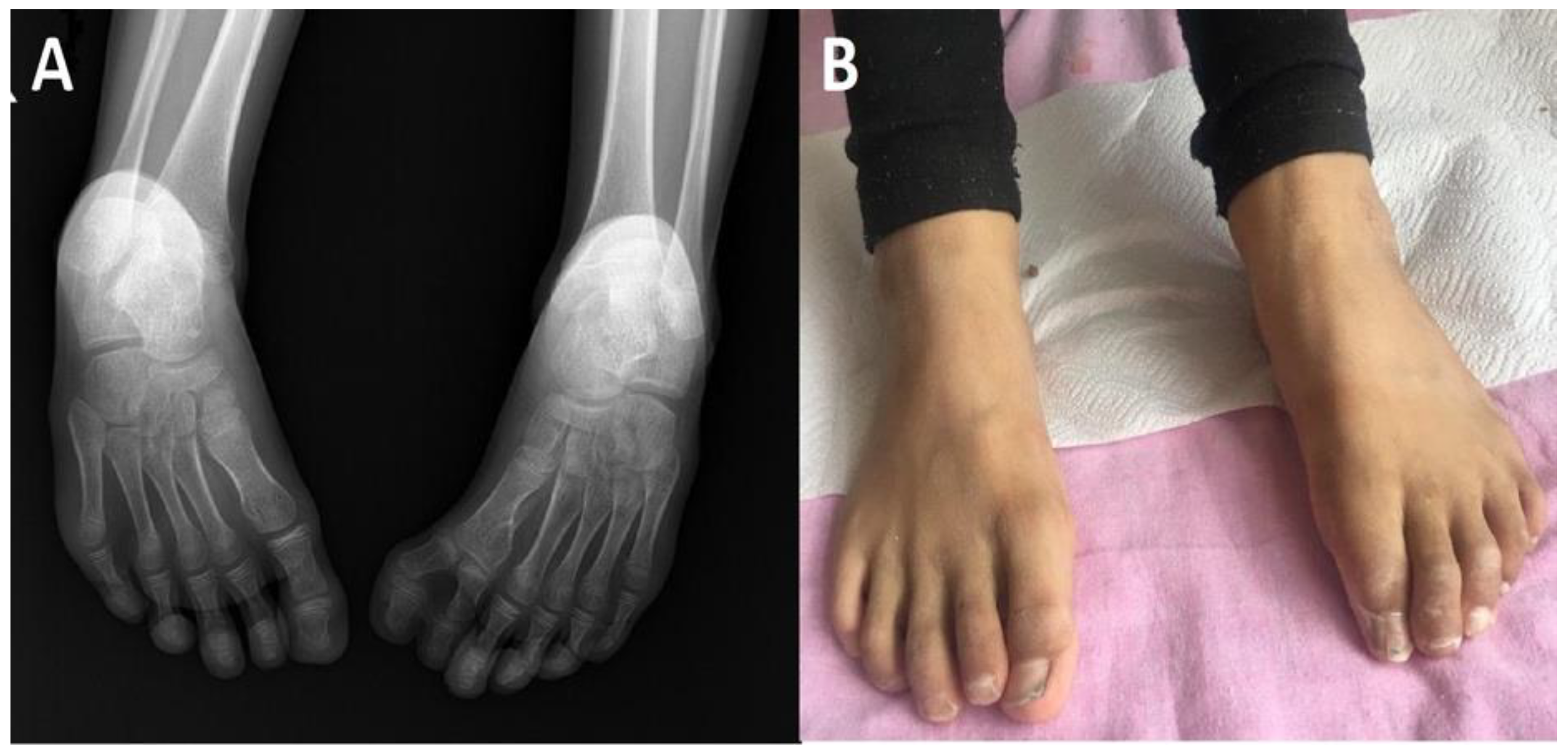{kind=link}
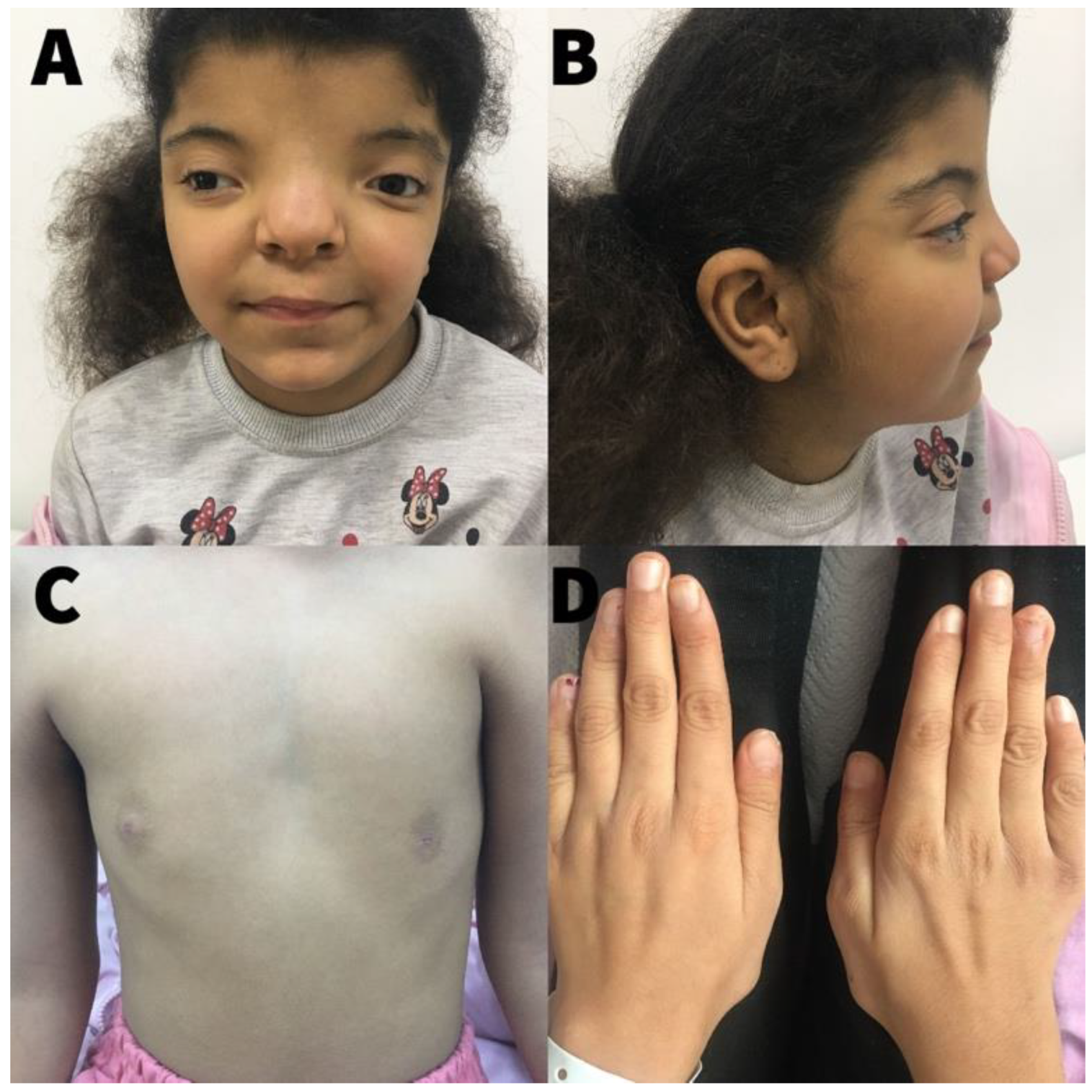{kind=link}
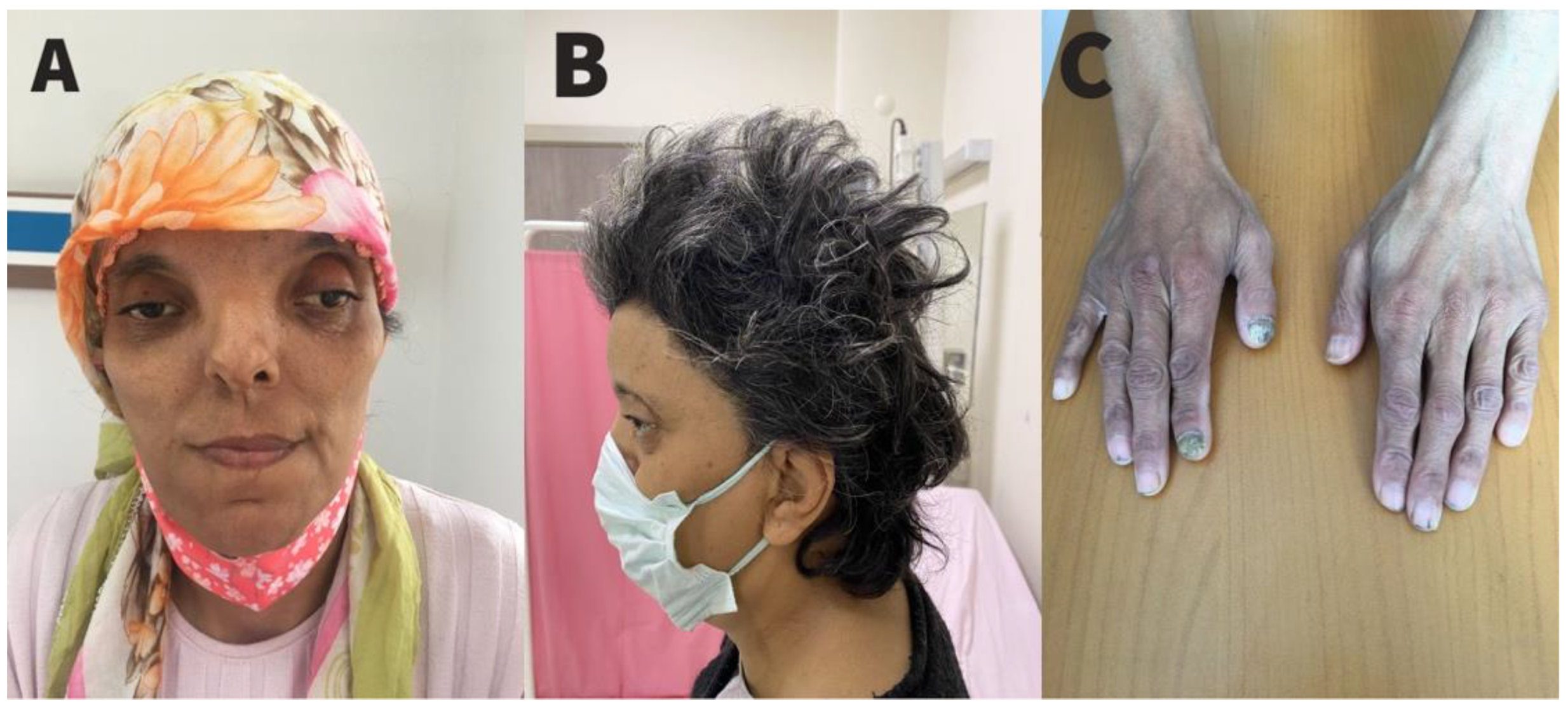{kind=link}
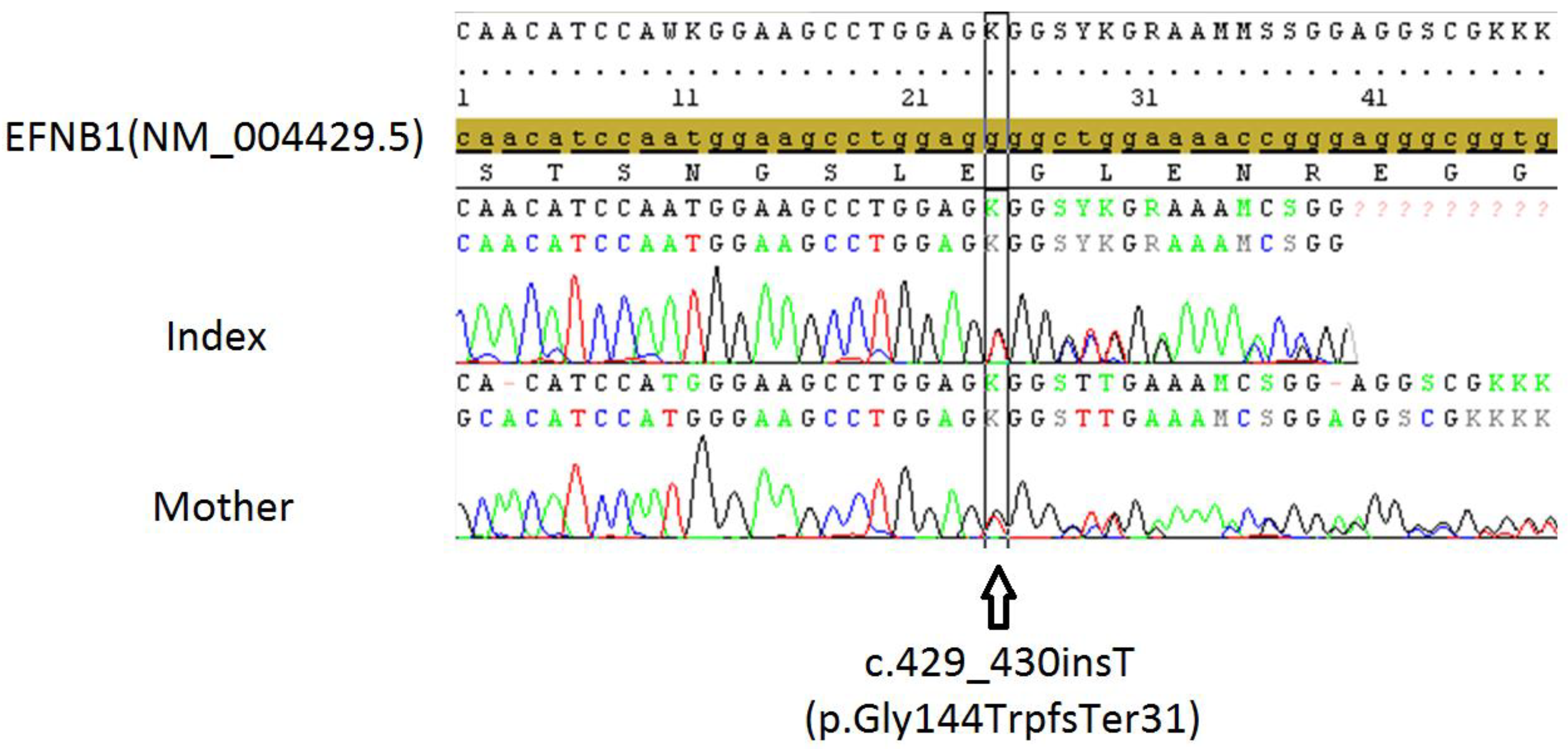{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Report
2.1. A General Description of the Patients
2.2. Face Anthropometry
2.3. Laboratory Findings
2.4. Genetic Tests
2.5. Screening for Comorbidities
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, M.M., Jr. Craniofrontonasal dysplasia. Birth Defects Orig. Artic. Ser. 1979, 15, 85–89. [Google Scholar] [PubMed]
- van den Elzen, M.E.; Twigg, S.R.; Goos, J.A.; Hoogeboom, A.J.; van den Ouweland, A.M.; Wilkie, A.O.; Mathijssen, I.M. Phenotypes of craniofrontonasal syndrome in patients with a pathogenic mutation in EFNB1. Eur. J. Hum. Genet. 2014, 22, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paddison, P.J.; Hannon, G.J. siRNAs and shRNAs: Skeleton keys to the human genome. Curr. Opin. Mol. Ther. 2003, 5, 217–224. [Google Scholar] [PubMed]
- Twigg, S.R.; Kan, R.; Babbs, C.; Bochukova, E.G.; Robertson, S.P.; Wall, S.A.; Morriss-Kay, G.M.; Wilkie, A.O. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8652–8657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Kong, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Darling, T.K.; Lamb, T.J. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Vail, M.E.; Ernst, M.; Scott, A.M. Eph Receptors in the Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 801–805. [Google Scholar] [CrossRef]
- Yu, G.; Luo, H.; Wu, Y.; Wu, J. EphrinB1 is essential in T-cell-T-cell co-operation during T-cell activation. J. Biol. Chem. 2004, 279, 55531–55539. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Qi, S.; Luo, H. The effect of conditional EFNB1 deletion in the T cell compartment on T cell development and function. BMC Immunol. 2011, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Mao, J.; Wu, Y.; Luo, H.; Wu, J. Ephrin-B1 is critical in T-cell development. J. Biol. Chem. 2006, 281, 10222–10229. [Google Scholar] [CrossRef]
- Wu, Z.; Luo, H.; Thorin, E.; Tremblay, J.; Peng, J.; Lavoie, J.L.; Wang, Y.; Qi, S.; Wu, T.; Wu, J. Possible Role of Efnb1 Protein, a Ligand of Eph Receptor Tyrosine Kinases, in Modulating Blood Pressure. J. Biol. Chem. 2012, 287, 15557–15569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wang, X.; Wu, Y.; Jin, W.; Cheng, B.; Fang, X.; Martel-Pelletier, J.; Kapoor, M.; Peng, J.; Qi, S.; et al. Role of EFNB1 and EFNB2 in Mouse Collagen-Induced Arthritis and Human Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Broux, B.; Wang, X.; Hu, Y.; Ghannam, S.; Jin, W.; Larochelle, C.; Prat, A.; Wu, J. EphrinB1 and EphrinB2 regulate T cell chemotaxis and migration in experimental autoimmune encephalomyelitis and multiple sclerosis. Neurobiol. Dis. 2016, 91, 292–306. [Google Scholar] [CrossRef]
- Luo, H.; Wu, Z.; Tremblay, J.; Thorin, E.; Peng, J.; Lavoie, J.L.; Hu, B.; Stoyanova, E.; Cloutier, G.; Qi, S. Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J. Biol. Chem. 2012, 287, 6819–6829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Wu, Z.; Qi, S.; Jin, W.; Han, B.; Wu, J. Ephrinb1 and Ephrinb2 are associated with interleukin-7 receptor alpha and retard its internalization from the cell surface. J. Biol. Chem. 2011, 286, 44976–44987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Shih, C.; Qi, H. Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science 2017, 356, eaai9264. [Google Scholar] [CrossRef]
- Mori, T.; Maeda, N.; Inoue, K.; Sekimoto, R.; Tsushima, Y.; Matsuda, K.; Yamaoka, M.; Suganami, T.; Nishizawa, H.; Ogawa, Y.; et al. A novel role for adipose ephrin-B1 in inflammatory response. PLoS ONE 2013, 8, e76199. [Google Scholar] [CrossRef]
- Twigg, S.R.; Matsumoto, K.; Kidd, A.M.; Goriely, A.; Taylor, I.B.; Fisher, R.B.; Hoogeboom, A.J.; Mathijssen, I.M.; Lourenco, M.T.; Morton, J.E.; et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: Frequent somatic mosaicism and explanation of the paucity of carrier males. Am. J. Hum. Genet. 2006, 78, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Compagni, A.; Logan, M.; Klein, R.; Adams, R.H. Control of skeletal patterning by ephrinB1-EphB interactions. Dev. Cell 2003, 5, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Luo, H.; Wu, Y.; Wu, J. Mouse ephrinB3 augments T-cell signaling and responses to T-cell receptor ligation. J. Biol. Chem. 2003, 278, 47209–47216. [Google Scholar] [CrossRef]
- Yu, G.; Luo, H.; Wu, Y.; Wu, J. Ephrin B2 Induces T Cell Costimulation. J. Immunol. 2003, 171, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Luo, H. Recent advances on T-cell regulation by receptor tyrosine kinases. Curr. Opin. Hematol. 2005, 12, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Segal, B.M. T(H)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 2011, 23, 707–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Wolf-Eichbaum, D.; Duinkerken, G.; Scherbaum, W.A.; Kolb, H.; Noordzij, J.G.; Roep, B.O. Development of type 1 diabetes despite severe hereditary B-cell deficiency. N. Engl. J. Med. 2001, 345, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.L.; Kawaguchi, Y.; Bennett, S.T.; Copeman, J.B.; Cordell, H.J.; Pritchard, L.E.; Reed, P.W.; Gough, S.C.; Jenkins, S.C.; Palmer, S.M.; et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature 1994, 371, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, M.; Towns, R.; Eisenbarth, G.S. Humoral autoimmunity in type 1 diabetes: Prediction, significance, and detection of distinct disease subtypes. Cold Spring Harb. Perspect. Med. 2012, 2, a012831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Xiu, Y.; Wong, C.P.; Bouaziz, J.-D.; Hamaguchi, Y.; Wang, Y.; Pop, S.M.; Tisch, R.M.; Tedder, T.F. B Lymphocyte Depletion by CD20 Monoclonal Antibody Prevents Diabetes in Nonobese Diabetic Mice despite Isotype-Specific Differences in FcγR Effector Functions. J. Immunol. 2008, 180, 2863–2875. [Google Scholar] [CrossRef] [Green Version]
- Weetman, A.P. An update on the pathogenesis of Hashimoto’s thyroiditis. J. Endocrinol. Investig. 2021, 44, 883–890. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, Q.; Wang, X.; Wang, S.; Shi, X.; Shan, Z.; Teng, W. Reversal of Abnormal CD4+ T Cell Metabolism Alleviates Thyroiditis by Deactivating the mTOR/HIF1a/Glycolysis Pathway. Front. Endocrinol. 2021, 12, 659738. [Google Scholar] [CrossRef]
- Dupond, J.L.; Essalmi, L.; Gil, H.; Meaux-Ruault, N.; Hafsaoui, C. Rituximab treatment of stiff-person syndrome in a patient with thymoma, diabetes mellitus and autoimmune thyroiditis. J. Clin. Neurosci. 2010, 17, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Anderson, M.S. Autoimmune Polyendocrine Syndromes: Clues to Type 1 Diabetes Pathogenesis. Immunity 2010, 32, 479–487. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güneş, S.; Wu, J.; Özyılmaz, B.; Deveci Sevim, R.; Ünüvar, T.; Anık, A. Cooccurring Type 1 Diabetes Mellitus and Autoimmune Thyroiditis in a Girl with Craniofrontonasal Syndrome: Are EFNB1 Variants Associated with Autoimmunity? Pharmaceuticals 2022, 15, 1535. https://doi.org/10.3390/ph15121535
Güneş S, Wu J, Özyılmaz B, Deveci Sevim R, Ünüvar T, Anık A. Cooccurring Type 1 Diabetes Mellitus and Autoimmune Thyroiditis in a Girl with Craniofrontonasal Syndrome: Are EFNB1 Variants Associated with Autoimmunity? Pharmaceuticals. 2022; 15(12):1535. https://doi.org/10.3390/ph15121535
Chicago/Turabian StyleGüneş, Sebla, Jiangping Wu, Berk Özyılmaz, Reyhan Deveci Sevim, Tolga Ünüvar, and Ahmet Anık. 2022. "Cooccurring Type 1 Diabetes Mellitus and Autoimmune Thyroiditis in a Girl with Craniofrontonasal Syndrome: Are EFNB1 Variants Associated with Autoimmunity?" Pharmaceuticals 15, no. 12: 1535. https://doi.org/10.3390/ph15121535