
Lysicamine Reduces Protein Kinase B (AKT) Activation and Promotes Necrosis in Anaplastic Thyroid Cancer
 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results
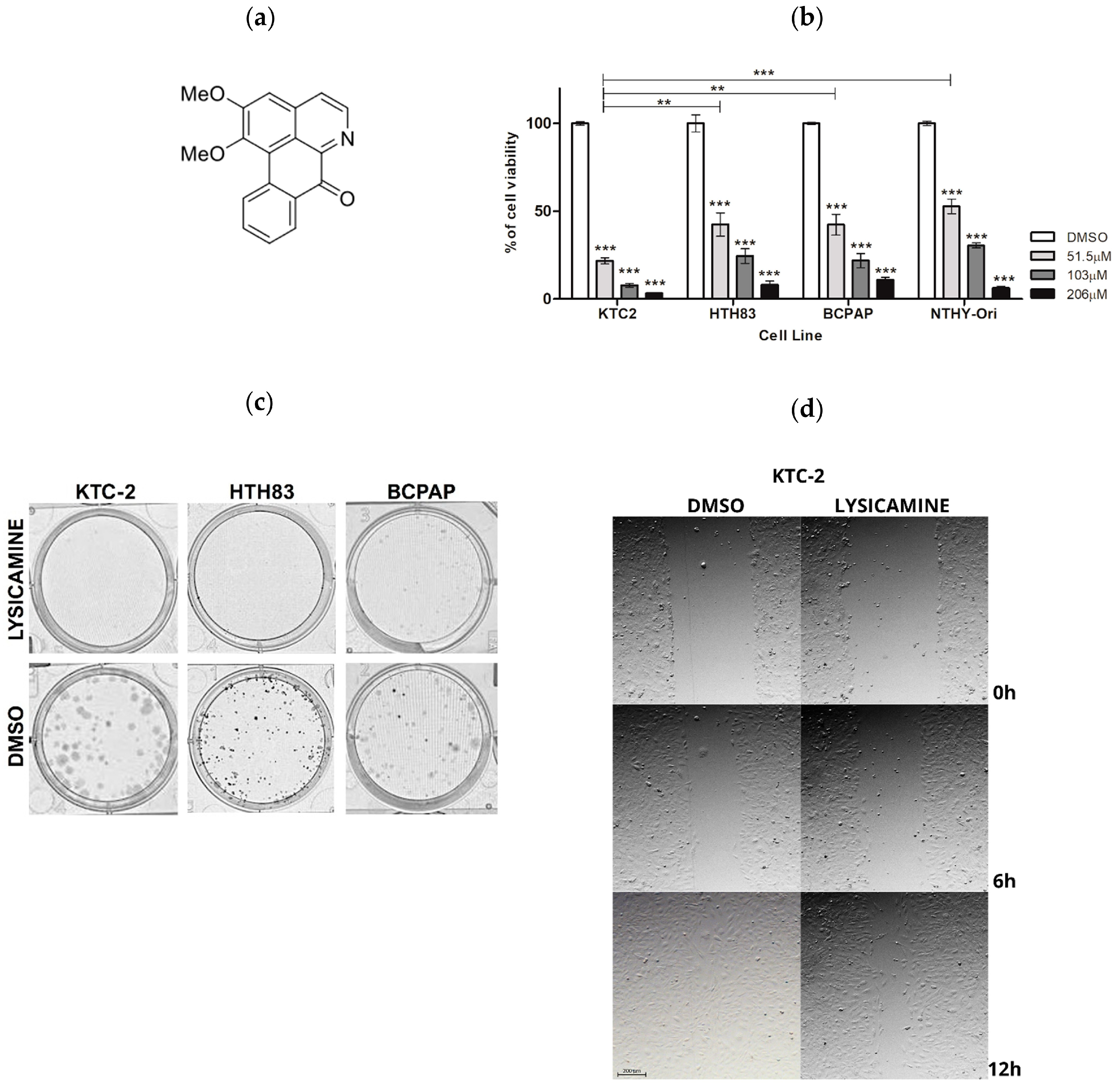
2.1. Lysicamine Induces Cell Toxicity in Monolayer Cell Culture
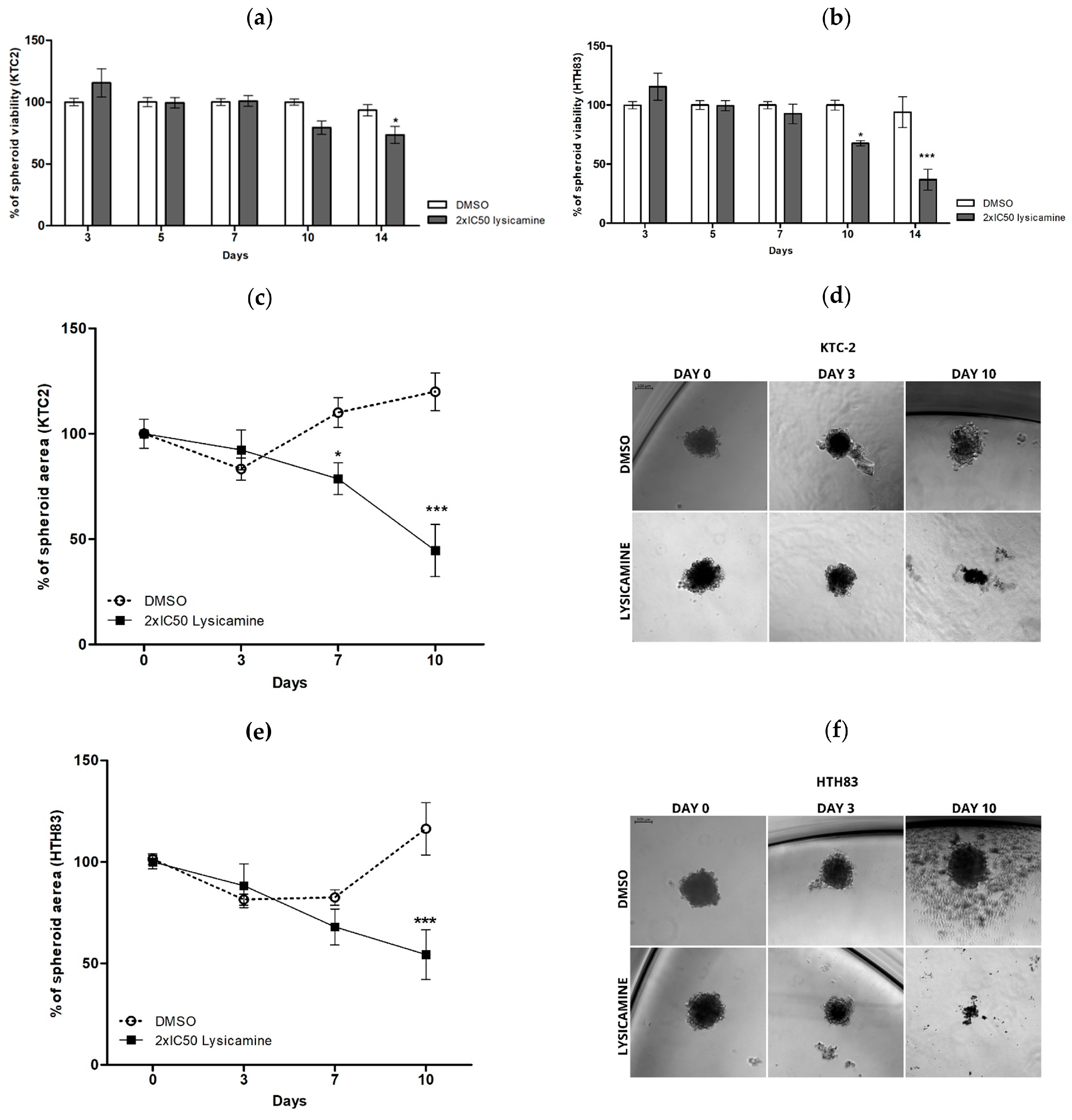
2.2. Lysicamine Induces Cell Toxicity in the Tumor Spheroids
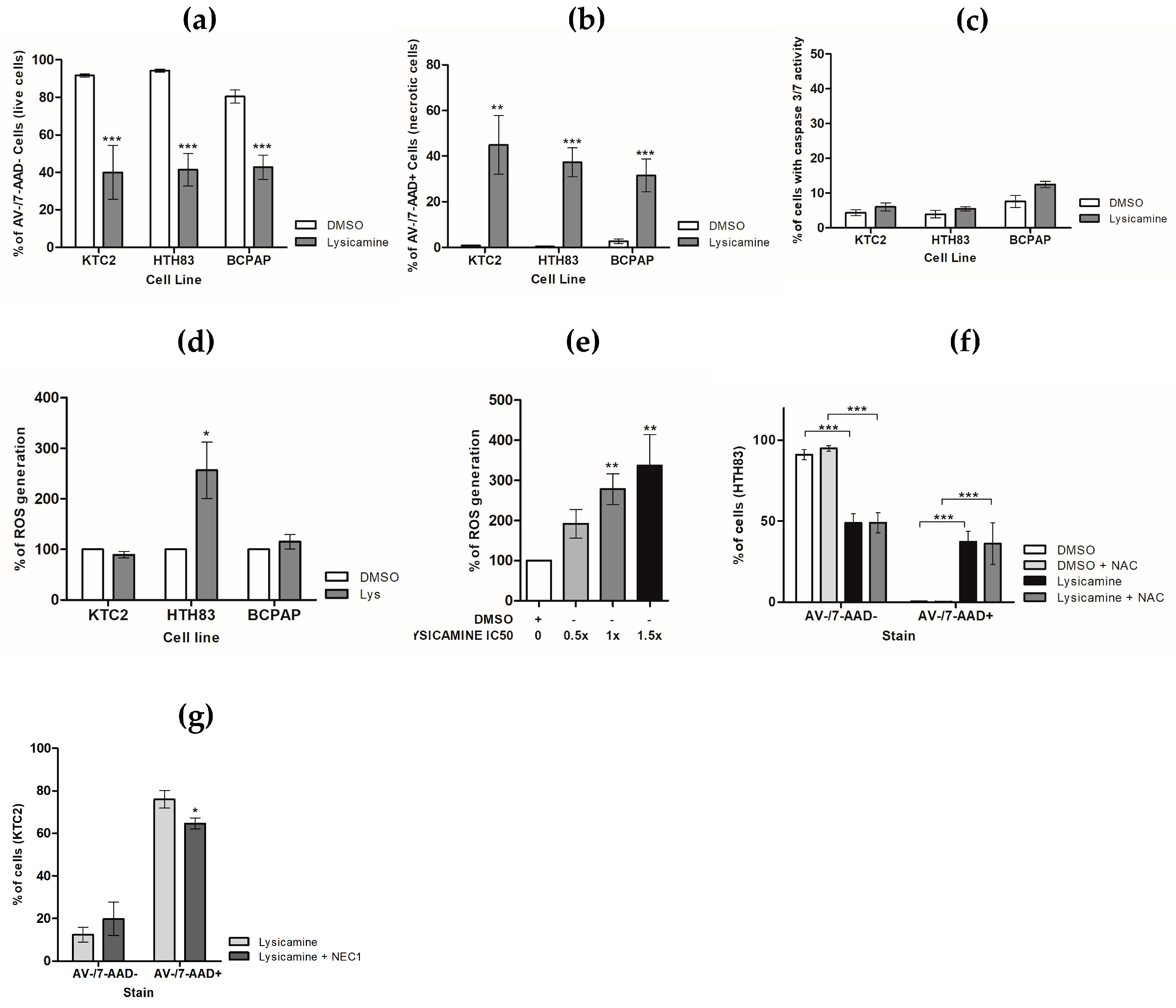
2.3. Lysicamine Promoted Necroptotic Cell Death in a Reactive Oxygen Species (ROS)-Independent Manner
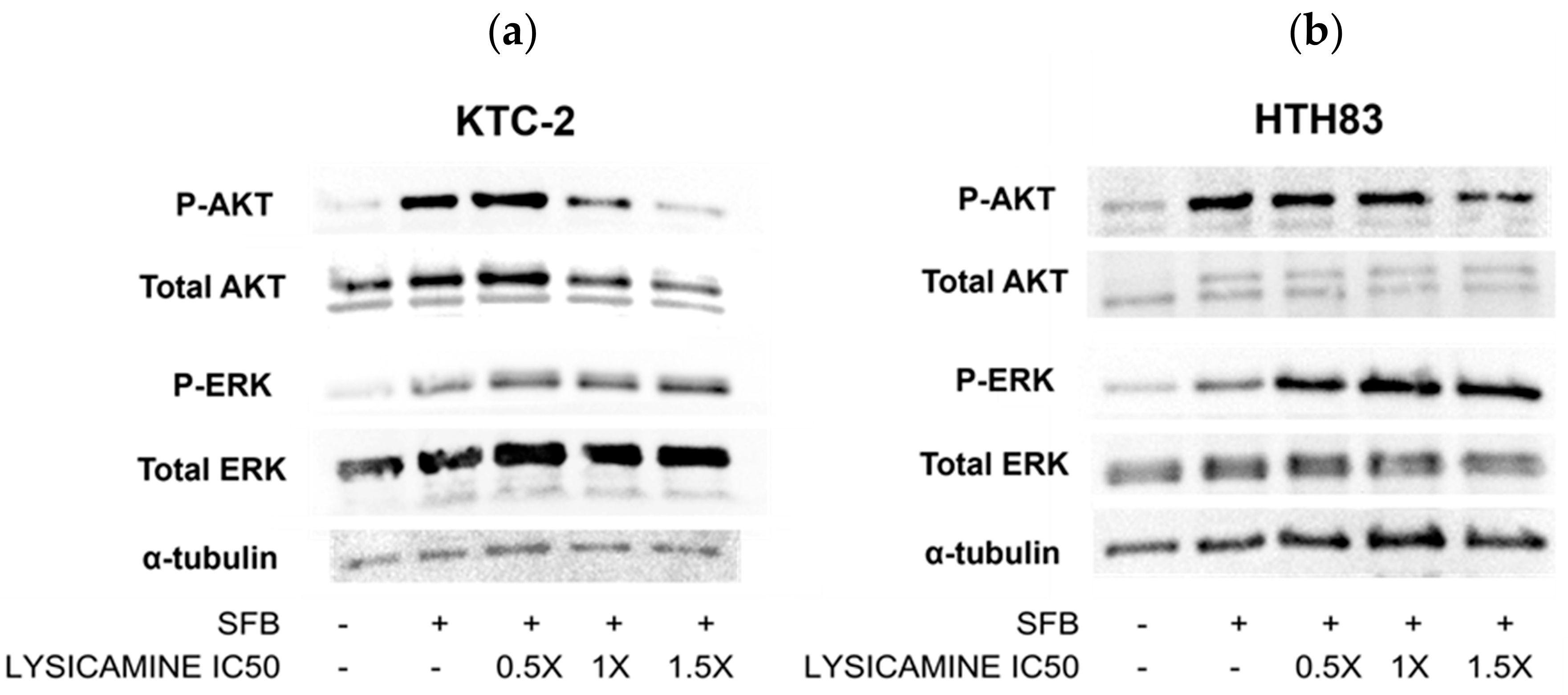
2.4. Lysicamine Modulates AKT Phosphorylation
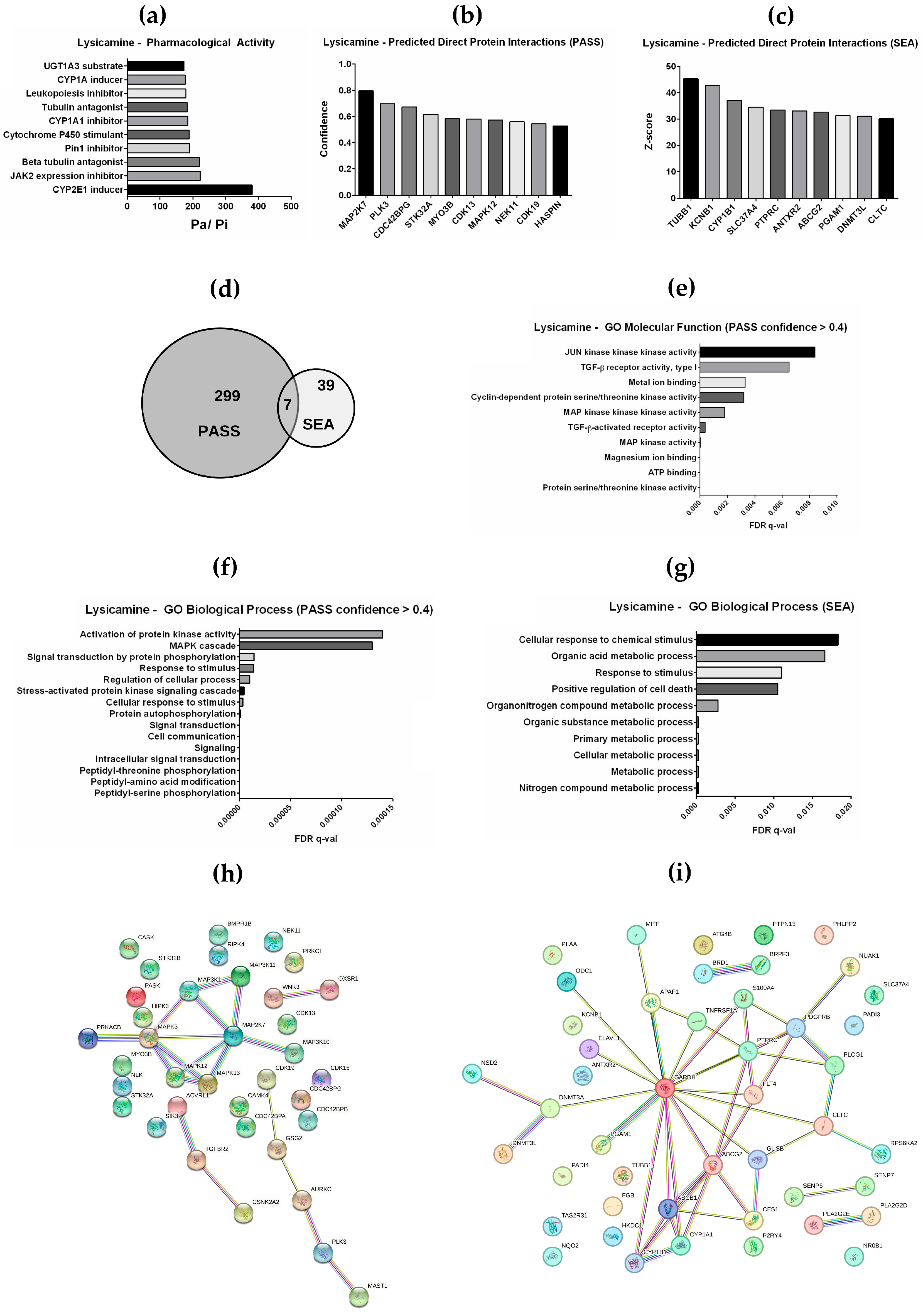
2.5. In Silico Prediction of Pharmacological Activity and Interacting Proteins for Lysicamine
3. Discussion
4. Materials and Methods
4.1. Cells, Reagents, and Treatments
4.2. Cell Viability, Proliferation, and IC50 and SI Determination
4.3. Clonogenic Assay
4.4. Cell Death Assays
4.5. Assessment of Reactive Oxygen Species (ROS)
4.6. Computational Target Prediction
4.7. AKT and ERK Pathways Analysis
4.8. Cell Migration
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- da Silva, J.A.G. Estimate/2016—Cancer Incidence in Brazil; INCA: Rio de Janeiro, Brazil, 2015; ISBN 978-85-7318-283-5. [Google Scholar]
- Instituto Nacional de Câncer. Estimate | 2023—Cancer Incidence in Brazil; Instituto Nacional de Câncer: Rio de Janeiro, Brazil, 2022; ISBN 978-65-88517-10-9. [Google Scholar]
- Li, M.; Brito, J.P.; Vaccarella, S. Long-Term Declines of Thyroid Cancer Mortality: An International Age–Period–Cohort Analysis. Thyroid 2020, 30, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Derwahl, M.; Nicula, D. Estrogen and Its Role in Thyroid Cancer. Endocr. Relat. Cancer 2014, 21, T273–T283. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S. Iodine Intake and Thyroid Carcinoma—A Potential Risk Factor. Exp. Clin. Endocrinol. Diabetes 2009, 106, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, F.S. Radiation Exposure and Thyroid Cancer. JAMA J. Am. Med. Assoc. 1977, 237, 2089. [Google Scholar] [CrossRef]
- Schmid, D.; Ricci, C.; Behrens, G.; Leitzmann, M.F. Adiposity and Risk of Thyroid Cancer: A Systematic Review and Meta-Analysis. Obes. Rev. 2015, 16, 1042–1054. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Mohammed, A.A.; EL-Shentenawy, A. Advanced Thyroid Cancers: New Era of Treatment. Med. Oncol. 2014, 31, 49. [Google Scholar] [CrossRef]
- McIver, B.; Hay, I.D.; Giuffrida, D.F.; Dvorak, C.E.; Grant, C.S.; Thompson, G.B.; van Heerden, J.A.; Goellner, J.R. Anaplastic Thyroid Carcinoma: A 50-Year Experience at a Single Institution. Surgery 2001, 130, 1028–1034. [Google Scholar] [CrossRef]
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic Thyroid Carcinoma: From Clinicopathology to Genetics and Advanced Therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Kim, M.; Jin, M.; Jeon, M.J.; Kim, E.Y.; Shin, D.Y.; Lim, D.J.; Kim, B.H.; Kang, H.-C.; Kim, W.B.; Shong, Y.K.; et al. Lenvatinib Compared with Sorafenib as a First-Line Treatment for Radioactive Iodine-Refractory, Progressive, Differentiated Thyroid Carcinoma: Real-World Outcomes in a Multicenter Retrospective Cohort Study. Thyroid 2023, 33, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET -Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.R.; Montierth, M.; Xu, L.; Goswami, M.; Zhao, X.; Cote, G.; Wang, W.; Iyer, P.; Dadu, R.; Busaidy, N.L.; et al. Impact of Somatic Mutations on Survival Outcomes in Patients With Anaplastic Thyroid Carcinoma. JCO Precis. Oncol. 2022, 6, e2100504. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Tsang, V.H.M.; Clifton-Bligh, R.J.; Robinson, B.G. Multikinase Inhibitors in Thyroid Cancer: Timing of Targeted Therapy. Nat. Rev. Endocrinol. 2021, 17, 225–234. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of Iodine-Refractory BRAF V600E-Mutant Metastatic Papillary Thyroid Cancer with Dabrafenib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1028–1035. [Google Scholar] [CrossRef]
- Wang, J.; Wong, Y.-K.; Liao, F. What Has Traditional Chinese Medicine Delivered for Modern Medicine? Expert Rev. Mol. Med. 2018, 20, e4. [Google Scholar] [CrossRef]
- Osorio, E.J.; Robledo, S.M.; Bastida, J. Chapter 2 Alkaloids with Antiprotozoal Activity. In The Alkaloids: Chemistry and Biology; Academic Press: Cambridge, MA, USA, 2008; pp. 113–190. [Google Scholar]
- Singh, I.P.; Bodiwala, H.S. Recent Advances in Anti-HIV Natural Products. Nat. Prod. Rep. 2010, 27, 1781. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Di, D.; Li, M.; Fen, Y. Structural and Mechanistic Bases of the Anticancer Activity of Natural Aporphinoid Alkaloids. Curr. Top. Med. Chem. 2013, 13, 2116–2126. [Google Scholar] [CrossRef]
- Omar, H.; Hashim, N.; Zajmi, A.; Nordin, N.; Abdelwahab, S.; Azizan, A.; Hadi, A.; Ali, H. Aporphine Alkaloids from the Leaves of Phoebe Grandis (Nees) Mer. (Lauraceae) and Their Cytotoxic and Antibacterial Activities. Molecules 2013, 18, 8994–9009. [Google Scholar] [CrossRef]
- Liu, C.M.; Kao, C.L.; Wu, H.M.; Li, W.J.; Huang, C.T.; Li, H.T.; Chen, C.Y. Antioxidant and Anticancer Aporphine Alkaloids from the Leaves of Nelumbo Nucifera Gaertn. Cv. Rosa-Plena. Molecules 2014, 19, 17829–17838. [Google Scholar] [CrossRef] [PubMed]
- Stevigny, C.; Bailly, C.; Quetin-Leclercq, J. Cytotoxic and Antitumor Potentialities of Aporphinoid Alkaloids. Curr. Med. Chem. -Anti-Cancer Agents 2005, 5, 173–182. [Google Scholar] [CrossRef]
- Cipriano Da Silva, T.R.; Oliveira, R.B.; Nicastro, A.L.M.; Ureshino, R.P.; Raminelli, C. Total Synthesis of Aporphine Alkaloids via Benzyne Chemistry: Progress Towards a Late-Stage Enantioselective Hydrogenation and Neuroprotective Activity Evaluations. ChemistrySelect 2023, 8, e202302821. [Google Scholar] [CrossRef]
- Rossini, A.F.C.; Muraca, A.C.A.; Casagrande, G.A.; Raminelli, C. Total Syntheses of Aporphine Alkaloids via Benzyne Chemistry: An Approach to the Formation of Aporphine Cores. J. Org. Chem. 2015, 80, 10033–10040. [Google Scholar] [CrossRef] [PubMed]
- Colella, G.; Fazioli, F.; Gallo, M.; De Chiara, A.; Apice, G.; Ruosi, C.; Cimmino, A.; de Nigris, F. Sarcoma Spheroids and Organoids—Promising Tools in the Era of Personalized Medicine. Int. J. Mol. Sci. 2018, 19, 615. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular Genetics and Diagnosis of Thyroid Cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating Protein Pharmacology by Ligand Chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- van Houten, P.; Netea-Maier, R.T.; Smit, J.W. Differentiated Thyroid Carcinoma: An Update. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 37, 101687. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Copland, J.A. Anaplastic Thyroid Carcinoma: Pathogenesis and Emerging Therapies. Clin. Oncol. 2010, 22, 486–497. [Google Scholar] [CrossRef]
- Prasongsook, N.; Kumar, A.; Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.; Molina, J.; Garces, Y.; Ma, D.; Wittich, M.A.N.; Rubin, J.; et al. Survival in Response to Multimodal Therapy in Anaplastic Thyroid Cancer. J. Clin. Endocrinol. Metab. 2017, 102, 4506–4514. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Meng, T.; Chen, Z.; Xie, X.; Qin, Q. Facile Total Synthesis of Lysicamine and the Anticancer Activities of the Ru II, Rh III, Mn II and Zn II Complexes of Lysicamine. Oncotarget 2017, 8, 59359–59375. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.F.; Liu, C.M.; Kao, C.L.; Chen, C.Y. Antioxidant and Anticancer Constituents from the Leaves of Liriodendron Tulipifera. Molecules 2014, 19, 4235–4245. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of In-Vitro Bioassay Methods: Application in Herbal Drug Research. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 46, pp. 273–307. ISBN 978-0-12-824127-1. [Google Scholar]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative Assay of 2D and 3D Cell Culture Models: Proliferation, Gene Expression and Anticancer Drug Response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Filipiak-Duliban, A.; Brodaczewska, K.; Kajdasz, A.; Kieda, C. Spheroid Culture Differentially Affects Cancer Cell Sensitivity to Drugs in Melanoma and RCC Models. Int. J. Mol. Sci. 2022, 23, 1166. [Google Scholar] [CrossRef] [PubMed]
- Leonard, F.; Godin, B. 3D In Vitro Model for Breast Cancer Research Using Magnetic Levitation and Bioprinting Method. In Breast Cancer; Cao, J., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1406, pp. 239–251. ISBN 978-1-4939-3442-3. [Google Scholar]
- Ware, M.J.; Colbert, K.; Keshishian, V.; Ho, J.; Corr, S.J.; Curley, S.A.; Godin, B. Generation of Homogenous Three-Dimensional Pancreatic Cancer Cell Spheroids Using an Improved Hanging Drop Technique. Tissue Eng.-Part C Methods 2016, 22, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Zoetemelk, M.; Rausch, M.; Colin, D.J.; Dormond, O.; Nowak-Sliwinska, P. Short-Term 3D Culture Systems of Various Complexity for Treatment Optimization of Colorectal Carcinoma. Sci. Rep. 2019, 9, 7103. [Google Scholar] [CrossRef]
- Perut, F.; Sbrana, F.V.; Avnet, S.; De Milito, A.; Baldini, N. Spheroid-Based 3D Cell Cultures Identify Salinomycin as a Promising Drug for the Treatment of Chondrosarcoma. J. Orthop. Res. 2018, 36, 2305–2312. [Google Scholar] [CrossRef]
- Malhão, F.; Ramos, A.A.; Macedo, A.C.; Rocha, E. Cytotoxicity of Seaweed Compounds, Alone or Combined to Reference Drugs, against Breast Cell Lines Cultured in 2D and 3D. Toxics 2021, 9, 24. [Google Scholar] [CrossRef]
- Tölle, R.C.; Gaggioli, C.; Dengjel, J. Three-Dimensional Cell Culture Conditions Affect the Proteome of Cancer-Associated Fibroblasts. J. Proteome Res. 2018, 17, 2780–2789. [Google Scholar] [CrossRef]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of Spherically Structured 3D in Vitro Tumor Models -Advances and Prospects. Acta Biomater. 2018, 75, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, J.K.; Hummon, A.B. Quantitative Evaluation of Liposomal Doxorubicin and Its Metabolites in Spheroids. Anal. Bioanal. Chem. 2019, 411, 7087–7094. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Lee, S.O. Apomorphine Suppresses TNF-α-Induced MMP-9 Expression and Cell Invasion through Inhibition of ERK/AP-1 Signaling Pathway in MCF-7 Cells. Biochem. Biophys. Res. Commun. 2017, 487, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.R.; Toker, A. Function of Akt/PKB Signaling to Cell Motility, Invasion and the Tumor Stroma in Cancer. Cell. Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Mortensen, A.C.; Haglöf, J.; Engskog, M.K.R.; Arvidsson, T.; Pettersson, C.; Glimelius, B.; Stenerlöw, B.; Nestor, M. Different Functions of AKT1 and AKT2 in Molecular Pathways, Cell Migration and Metabolism in Colon Cancer Cells. Int. J. Oncol. 2017, 50, 5–14. [Google Scholar] [CrossRef]
- Torre, L.D.; Nebbioso, A.; Stunnenberg, H.G.; Martens, J.H.A.; Carafa, V.; Altucci, L. The Role of Necroptosis: Biological Relevance and Its Involvement in Cancer. Cancers 2021, 13, 684. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-Cell-Induced Endothelial Cell Necroptosis via Death Receptor 6 Promotes Metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Lim, J.W.; Kim, H. Astaxanthin Induces NADPH Oxidase Activation and Receptor-interacting Protein Kinase 1-mediated Necroptosis in Gastric Cancer AGS Cells. Mol. Med. Rep. 2021, 24, 837. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, P.; Shi, J.; Ji, X.; He, L.; Dong, W.; Wang, Z.; Zhang, H.; Sun, W. A Novel Necroptosis-Related Gene Signature Associated with Immune Landscape for Predicting the Prognosis of Papillary Thyroid Cancer. Front. Genet. 2022, 13, 947216. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant Responses and Cellular Adjustments to Oxidative Stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, C.; Mo, Y.-Y.; Hebbar, V.; Owuor, E.D.; Tan, T.-H.; Kong, A.-N.T. Activation of Mitogen-Activated Protein Kinase Pathways Induces Antioxidant Response Element-Mediated Gene Expression via a Nrf2-Dependent Mechanism. J. Biol. Chem. 2000, 275, 39907–39913. [Google Scholar] [CrossRef] [PubMed]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF -Mutant Melanoma to BRAF Inhibition Is Mediated by a Network of Transcriptional Regulators of Glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS Production Induced by BRAF Inhibitor Treatment Rewires Metabolic Processes Affecting Cell Growth of Melanoma Cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef]
- Zaballos, M.A.; Santisteban, P. Key Signaling Pathways in Thyroid Cancer. J. Endocrinol. 2017, 235, R43–R46. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Untch, B.R.; Xu, B.; Ghossein, R.; Han, C.; Kuo, F.; Valero, C.; Nadeem, Z.; Patel, N.; Makarov, V.; et al. Genomic and Transcriptomic Correlates of Thyroid Carcinoma Evolution after BRAF Inhibitor Therapy. Mol. Cancer Res. 2022, 20, 45–55. [Google Scholar] [CrossRef]
- Yoo, S.-K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.; Cho, S.W.; Won, J.-K.; et al. Integrative Analysis of Genomic and Transcriptomic Characteristics Associated with Progression of Aggressive Thyroid Cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Chung, Y.J.; Saji, M.; Ringel, M.D. AKT in Thyroid Tumorigenesis and Progression. Endocrinology 2007, 148, 942–947. [Google Scholar] [CrossRef]
- Silaghi, H.; Lozovanu, V.; Georgescu, C.E.; Pop, C.; Nasui, B.A.; Cătoi, A.F.; Silaghi, C.A. State of the Art in the Current Management and Future Directions of Targeted Therapy for Differentiated Thyroid Cancer. Int. J. Mol. Sci. 2022, 23, 3470. [Google Scholar] [CrossRef]
- Jin, N.; Jiang, T.; Rosen, D.M.; Nelkin, B.D.; Ball, D.W. Synergistic Action of a RAF Inhibitor and a Dual PI3K/mTOR Inhibitor in Thyroid Cancer. Clin. Cancer Res. 2011, 17, 6482–6489. [Google Scholar] [CrossRef]
- Yi, H.; Ye, X.; Long, B.; Ye, T.; Zhang, L.; Yan, F.; Yang, Y.; Li, L. Inhibition of the AKT/mTOR Pathway Augments the Anticancer Effects of Sorafenib in Thyroid Cancer. Cancer Biother. Radiopharm. 2017, 32, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Di Cristofano, F.; Ranieri, M.; De Martino, D.; Di Cristofano, A. PI3K/mTOR Inhibition Potentiates and Extends Palbociclib Activity in Anaplastic Thyroid Cancer. Endocr.-Relat. Cancer 2019, 26, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.-K.; Cui, W. PI3K/mTORC2 Regulates TGF-β/Activin Signalling by Modulating Smad2/3 Activity via Linker Phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef] [PubMed]
- Luckett, K.A.; Cracchiolo, J.R.; Krishnamoorthy, G.P.; Leandro-Garcia, L.J.; Nagarajah, J.; Saqcena, M.; Lester, R.; Im, S.Y.; Zhao, Z.; Lowe, S.W.; et al. Co-Inhibition of SMAD and MAPK Signaling Enhances 124I Uptake in BRAF-Mutant Thyroid Cancers. Endocr.-Relat. Cancer 2021, 28, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Cuny, G.D.; Degterev, A. RIPK Protein Kinase Family: Atypical Lives of Typical Kinases. Semin. Cell Dev. Biol. 2021, 109, 96–105. [Google Scholar] [CrossRef]
- Madej, E.; Brożyna, A.A.; Adamczyk, A.; Wronski, N.; Harazin-Lechowska, A.; Muzyk, A.; Makuch, K.; Markiewicz, M.; Rys, J.; Wolnicka-Glubisz, A. Vemurafenib and Dabrafenib Downregulates RIPK4 Level. Cancers 2023, 15, 918. [Google Scholar] [CrossRef]
- Yi, H.; Su, Y.; Lin, R.; Zheng, X.; Pan, D.; Lin, D.; Gao, X.; Zhang, R. Downregulation of RIPK4 Expression Inhibits Epithelial-Mesenchymal Transition in Ovarian Cancer through IL-6. J. Immunol. Res. 2021, 2021, 8875450. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, F.; Hong, C.-Q.; Giuliano, A.E.; Cui, X.-J.; Zhou, G.-J.; Zhang, G.-J.; Cui, Y.-K. Critical Protein GAPDH and Its Regulatory Mechanisms in Cancer Cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. High Iodine Blocks a Notch/miR-19 Loop Activated by the BRAF V600E Oncoprotein and Restores the Response to TGFβ in Thyroid Follicular Cells. Thyroid 2014, 24, 453–462. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Raminelli, C.; Hassanie, H.; Trossini, G.H.G.; Perecim, G.P.; Caja, L.; Moustakas, A.; Vettore, A.L. Aporphine and Isoquinoline Derivatives Block Glioblastoma Cell Stemness and Enhance Temozolomide Cytotoxicity. Sci. Rep. 2022, 12, 21113. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Lysicamine IC50 (µM) ± SD | Selectivity Index (SI) |
|---|---|---|
| KTC-2 (ATC) | 15.6 ± 2.1 | 1.98 |
| HTH83 (ATC) | 36.4 ± 11.6 | 0.85 |
| BCPAP (PTC) | 30.5 ± 13.2 | 1.01 |
| NThy-ORI (non-tumoral) | 30.9 ± 4.8 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, M.T.; Michelli, A.P.P.; Caso, G.F.; de Oliveira, P.R.; Rodrigues-Junior, D.M.; Morale, M.G.; Machado Júnior, J.; Bortoluci, K.R.; Tamura, R.E.; da Silva, T.R.C.; et al. Lysicamine Reduces Protein Kinase B (AKT) Activation and Promotes Necrosis in Anaplastic Thyroid Cancer. Pharmaceuticals 2023, 16, 1687. https://doi.org/10.3390/ph16121687
Rodrigues MT, Michelli APP, Caso GF, de Oliveira PR, Rodrigues-Junior DM, Morale MG, Machado Júnior J, Bortoluci KR, Tamura RE, da Silva TRC, et al. Lysicamine Reduces Protein Kinase B (AKT) Activation and Promotes Necrosis in Anaplastic Thyroid Cancer. Pharmaceuticals. 2023; 16(12):1687. https://doi.org/10.3390/ph16121687
Chicago/Turabian StyleRodrigues, Mariana Teixeira, Ana Paula Picaro Michelli, Gustavo Felisola Caso, Paloma Ramos de Oliveira, Dorival Mendes Rodrigues-Junior, Mirian Galliote Morale, Joel Machado Júnior, Karina Ramalho Bortoluci, Rodrigo Esaki Tamura, Tamiris Reissa Cipriano da Silva, and et al. 2023. "Lysicamine Reduces Protein Kinase B (AKT) Activation and Promotes Necrosis in Anaplastic Thyroid Cancer" Pharmaceuticals 16, no. 12: 1687. https://doi.org/10.3390/ph16121687