


Identification of Potential Multitarget Compounds against Alzheimer’s Disease through Pharmacophore-Based Virtual Screening

 , ,
, ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
1.1. Pathogenesis of Alzheimer’s Disease
1.2. Current Pharmacotherapy for Alzheimer’s Disease
1.3. Multitarget Inhibitors
2. Results and Discussion
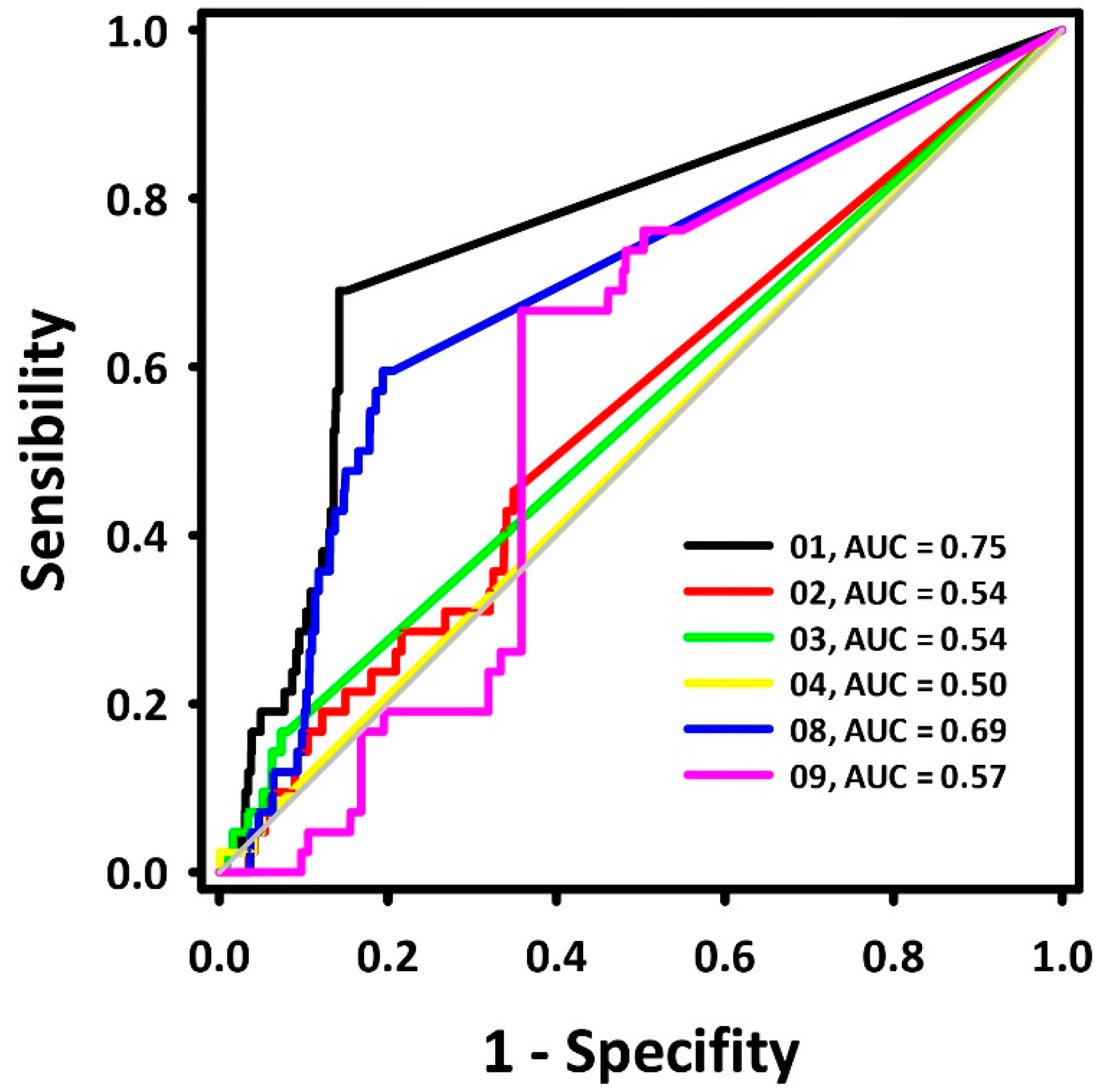
2.1. Pharmacophore Model Generation and Evaluation
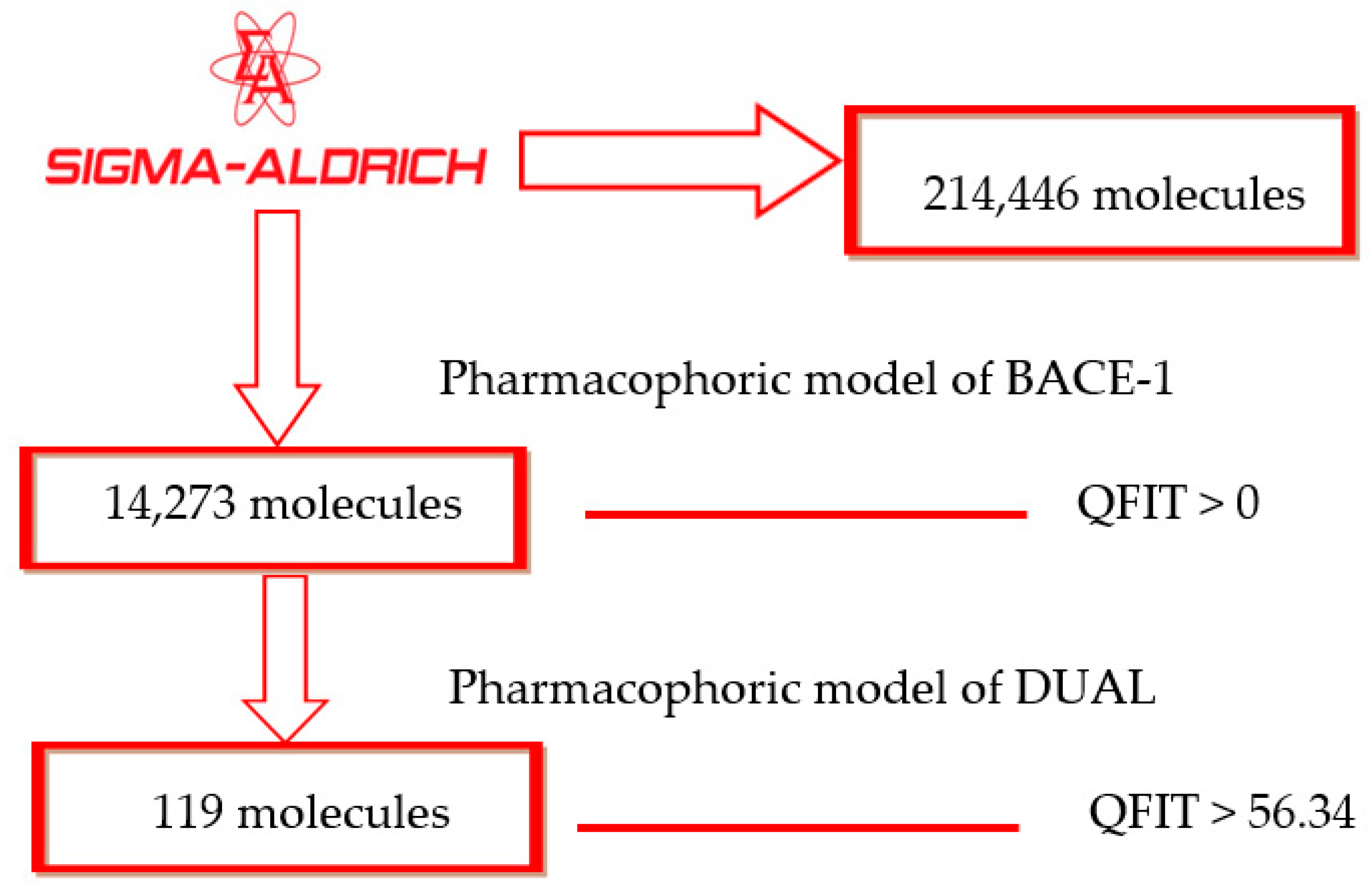
2.2. Pharmacophore-Based Virtual Screening
2.3. Molecular-Docking-Based Virtual Screening
2.4. Application of Physicochemical Filters
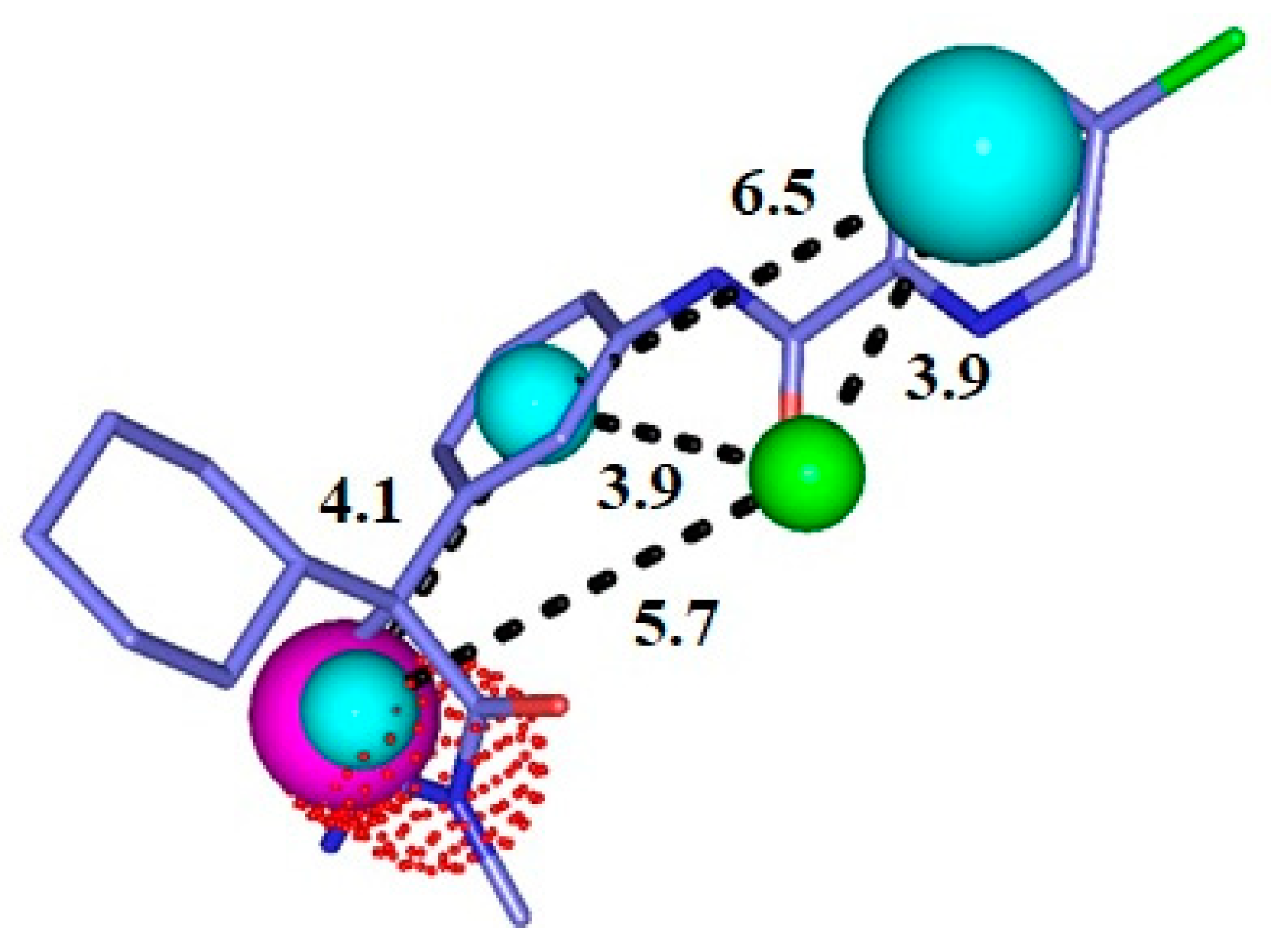
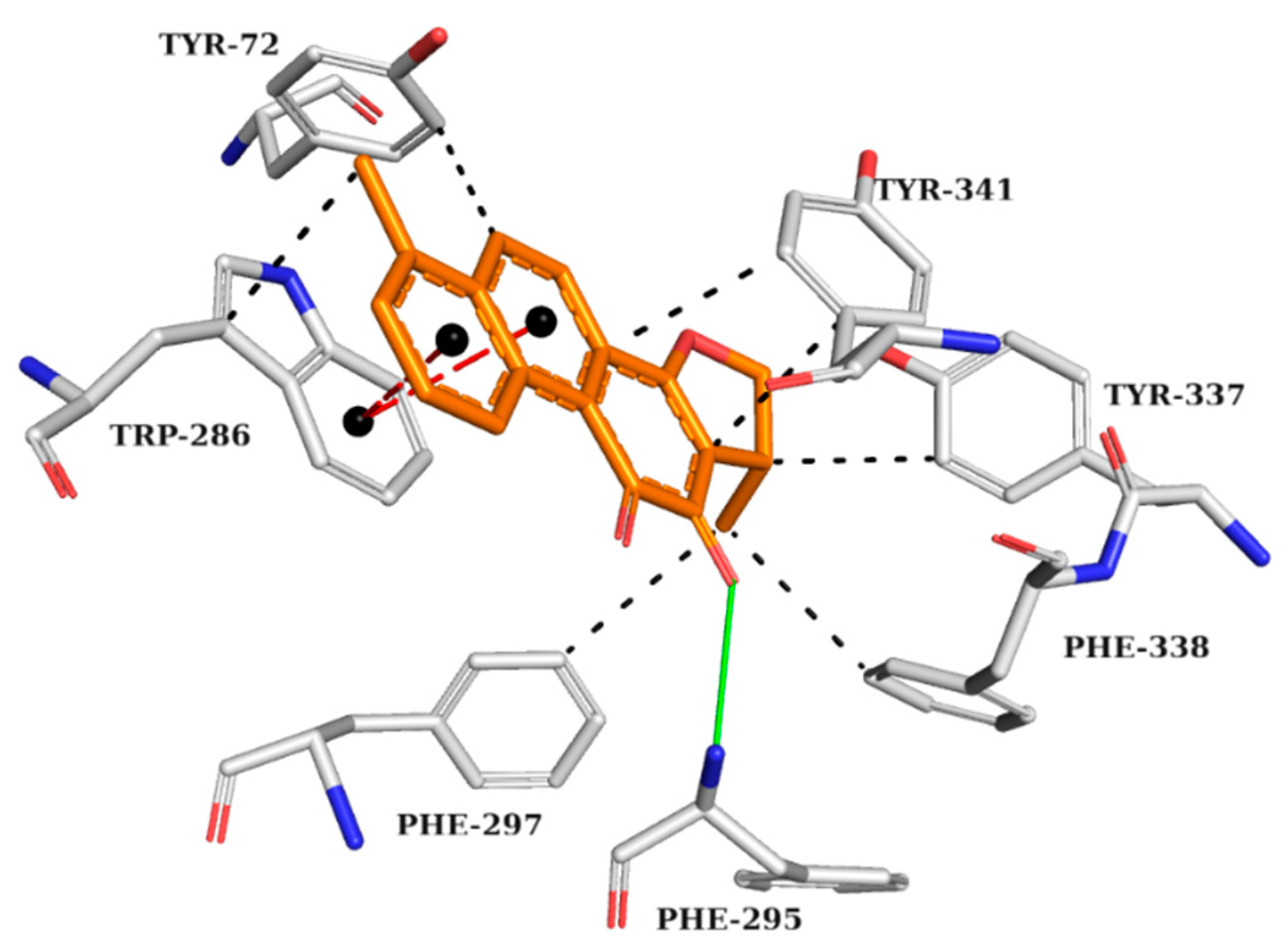
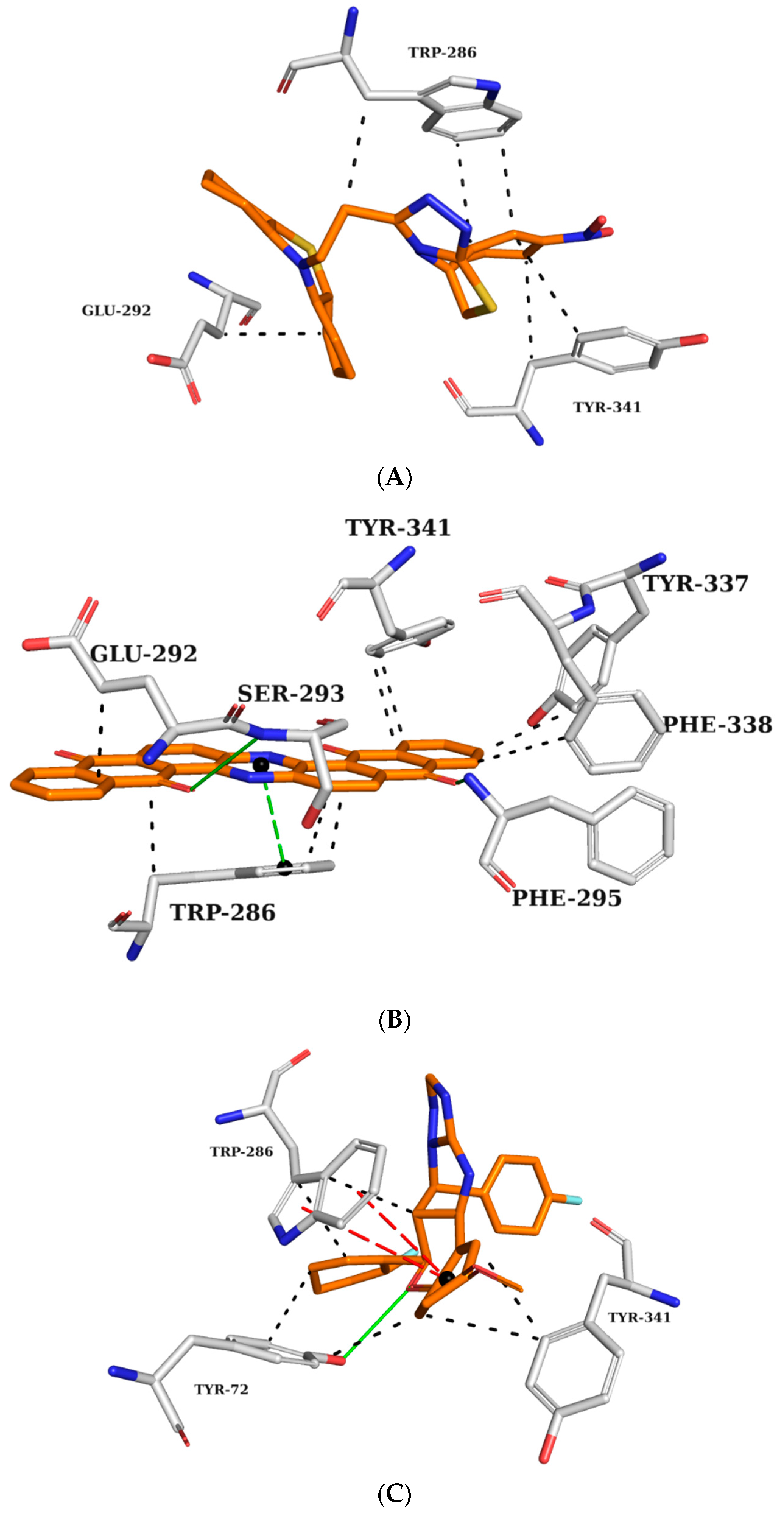
2.5. Analysis of Intermolecular Interactions
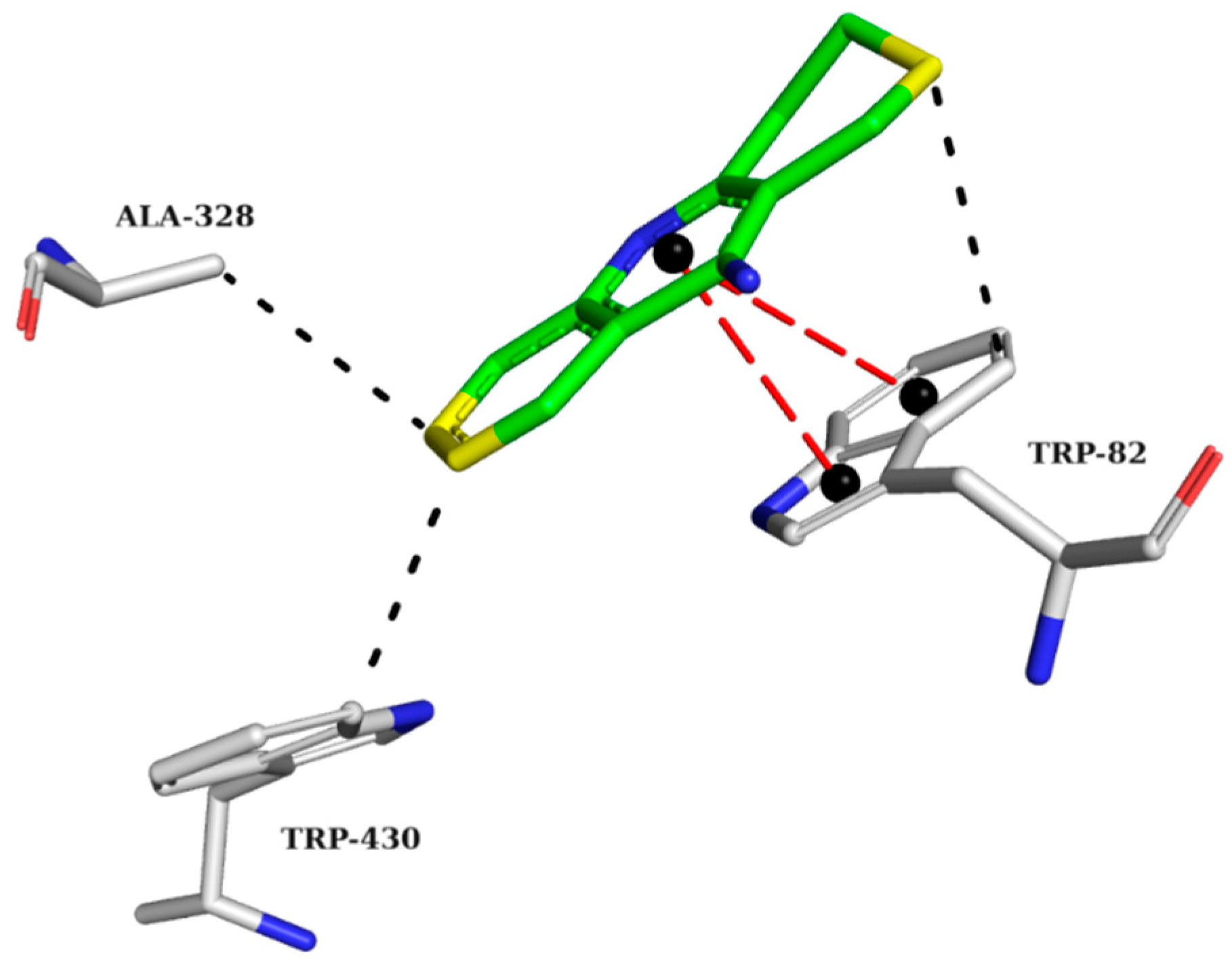
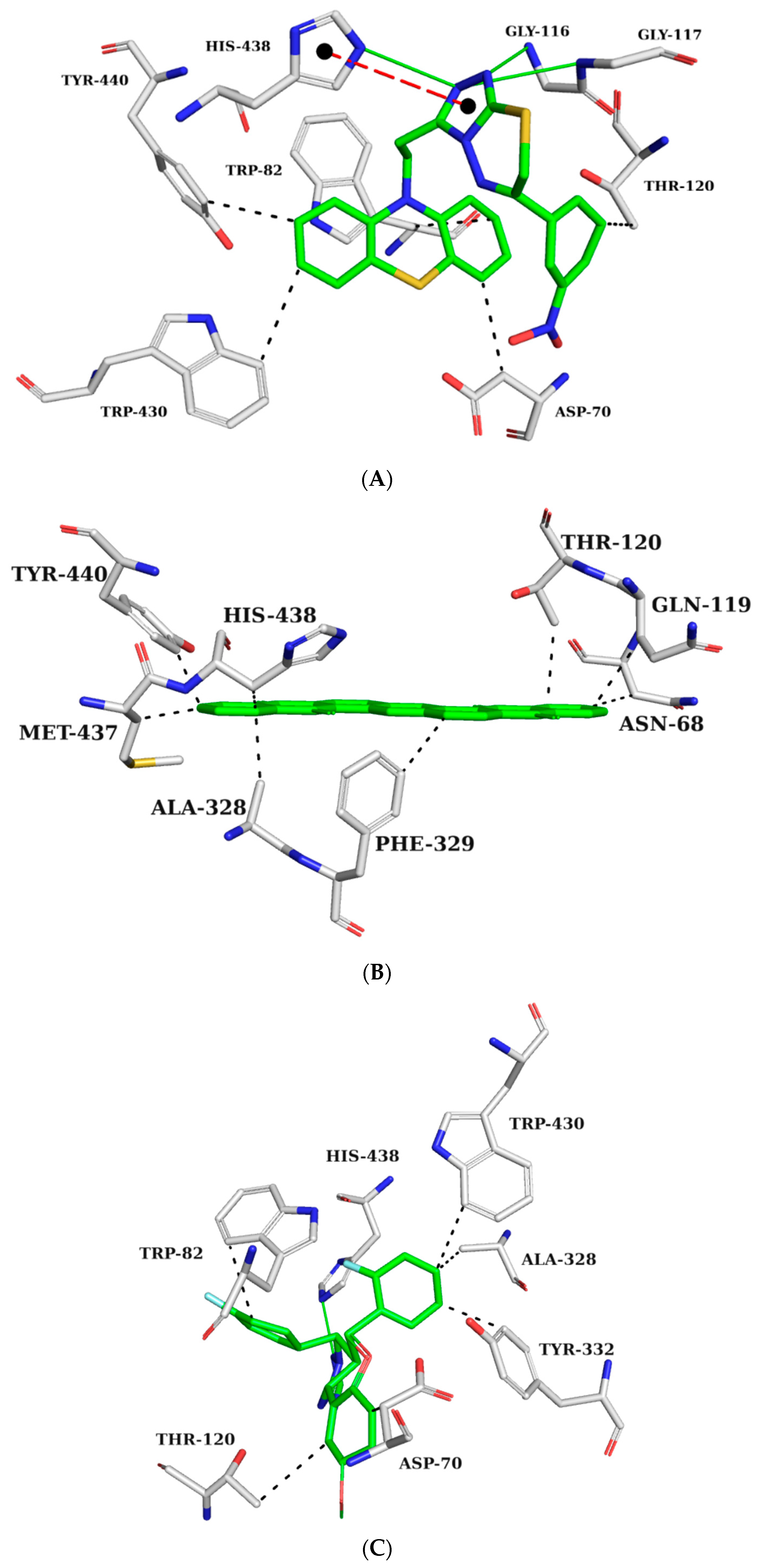
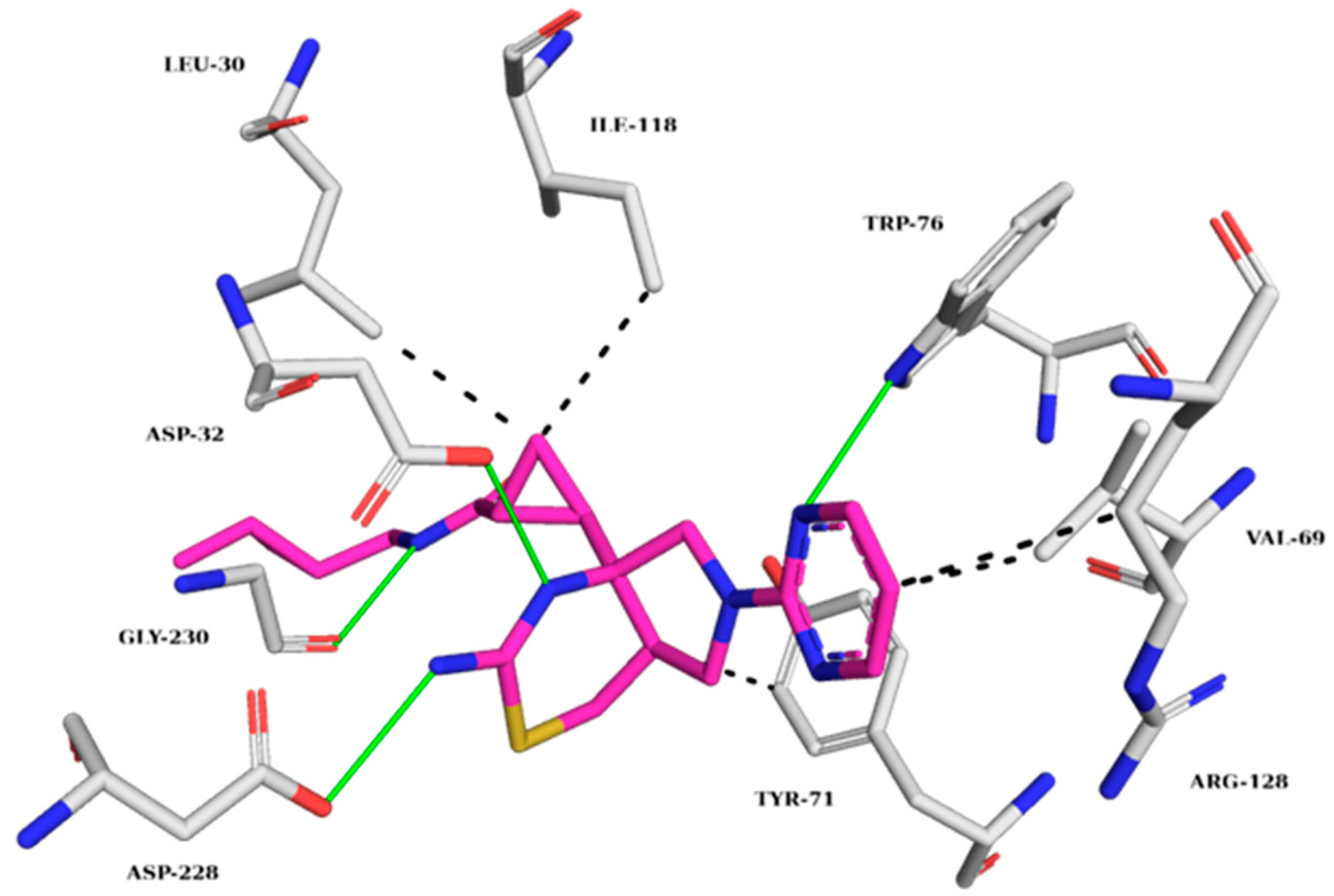
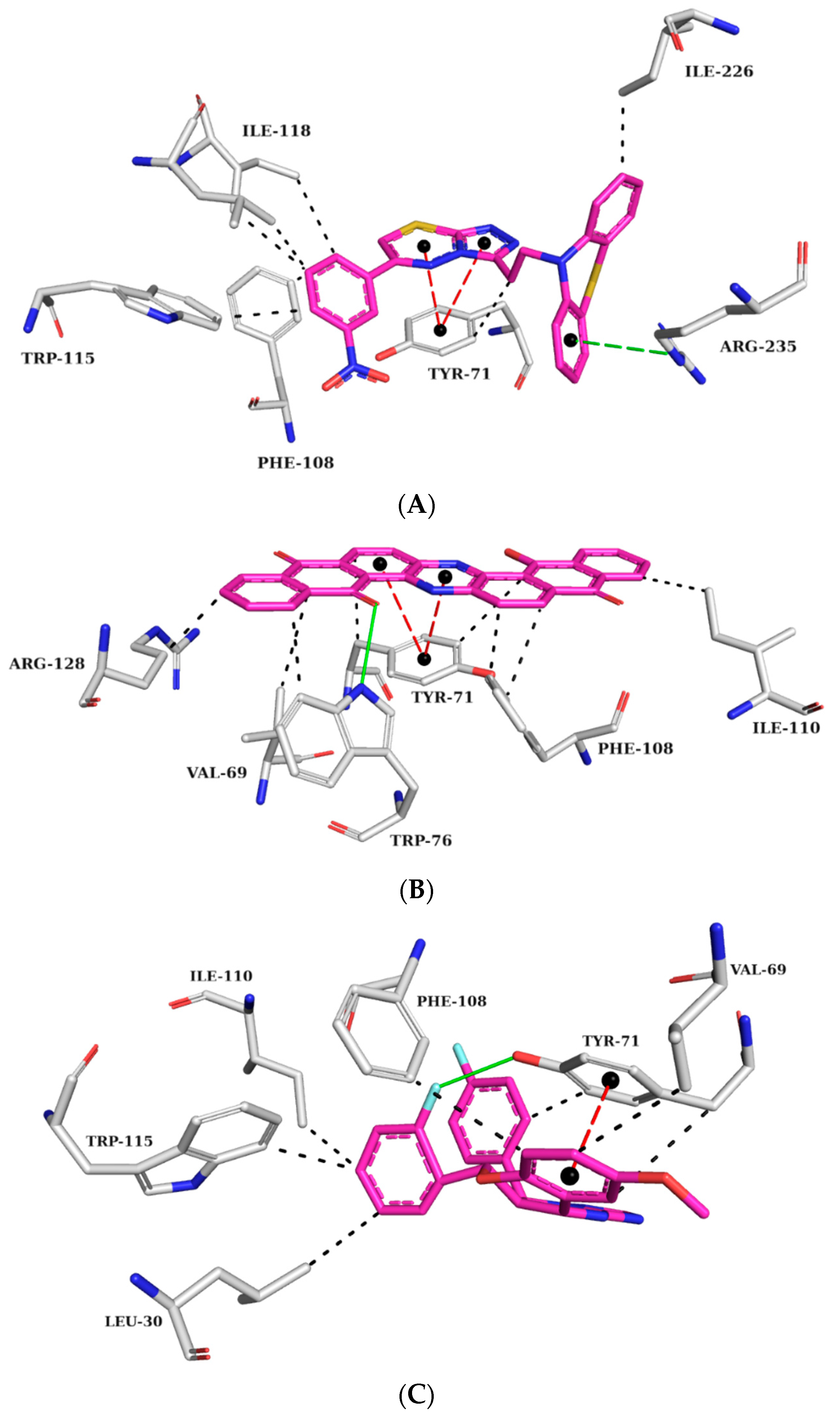
2.5.1. AChE Complexes
2.5.2. BChE Complexes
2.5.3. BACE-1 Complexes
2.6. Analysis of AMES Test (Cytotoxicity) and Other Parameters of Toxicity
3. Materials and Methods
3.1. Dataset
3.2. Pharmacophore Model Generation
3.3. Pharmacophore Model Evaluation
3.4. Pharmacophore-Based Virtual Screening
3.5. Molecular Docking
3.6. Physicochemical Filters
- Lipinski’s: Molecular Weight (MW) ≤ 500 Da; Hydrogen Bond Donors (HBD) ≤ 5; Hydrogen Bond Acceptors (HBA) ≤ 10; and cLogp ≤ 5.
- Veber’s: Polar Surface Area (PSA) ≤ 140 ; Rotatable Bonds (RB) ≤ 10; Sum of HBD and HBA ≤ 12.
3.7. Evaluation of Intermolecular Interactions
3.8. AMES Test (Cytotoxicity) and Other Parameters of Toxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Falco, A.; Cukierman, D.S.; Hauser-Davis, R.A.; Rey, N.A. Doença de Alzheimer: Hipóteses etiológicas e perspectivas de tratamento. Química Nova 2016, 39, 63–80. [Google Scholar] [CrossRef]
- World Health Organization. Dementia. [S. L.]; WHO: Geneva, Switzerland, 2019; Available online: https://www.who.int/es/news-room/fact-sheets/detail/dementia (accessed on 5 January 2020).
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Najafi, Z.; Mahdavi, M.; Saledi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Shariffadeh, M.; Akbarzadeh, T. Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational design of multitarget-directed ligands: Strategies and emerging paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef]
- Dias, K.S.T.; de Paula, C.T.; Riquiel, M.M.; Lago, S.T.; Costa, K.C.M.; Vaz, S.M.; Machado, R.P.; Lima, L.M.S.; Viegas Junior, C. Aplicações recentes da abordagem de fármacos multialvo para o tratamento da Doença de Alzheimer. Virtual Química 2015, 7, 609–648. [Google Scholar] [CrossRef]
- Mascarenhas, A.M.S.; de Almeida, R.B.M.; Araujo, M.F.N.; Mendes, G.O.; da Cruz, J.N.; dos Santos, C.B.R.; Botura, M.B.; Leite, F.H.A. Pharmacophore-based virtual screening and molecular docking to identify promising dual inhibitors of human acetylcholinesterase and butyrylcholinesterase. J. Biomol. Struct. Dyn. 2020, 39, 6021–6030. [Google Scholar] [CrossRef]
- Rodrigues, R.P.; Mantoani, S.P.; de Almeida, J.R.; Pinseta, F.R.; Semighini, E.P.; da Silva, V.B.; da Silva, C.H.P. Estratégias de Triagem Virtual no Planejamento de Fármacos. Rev. Virtual Química 2012, 4, 739–776. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mangold, M.; Howard, N.I.; Robinson, R.L.M.; Abell, C.; Blumberger, J.; Mitchell, J.B. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. 2012, 9, 3196–3207. [Google Scholar]
- Liu, K.P.Y.; Chan, C.C.H.; Chu, M.M.L.; Ng, T.Y.L.; Chu, L.W.; Hui, F.S.L.; Yuen, H.K.; Fisher, A.G. Activities of daily living performance in dementia. Acta Neurol. Scand. 2007, 116, 91–95. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J. Zinc 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar]
- Clark, R.D.; Abrahamian, E. Using a staged multi-objective optimization approach to find selective pharmacophore models. J. Comput. -Aided Mol. Des. 2009, 23, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Shepphird, J.K.; Clark, R.D. A marriage made in torsional space using GALAHAD models to drive pharmacophore multiplet searches. J. Comput. -Aided Mol. Des. 2006, 20, 763–771. [Google Scholar] [CrossRef]
- Metz, C.E. Basic principles of ROC analysis. Semin. Nucl. Med. 1978, 8, 283–298. [Google Scholar]
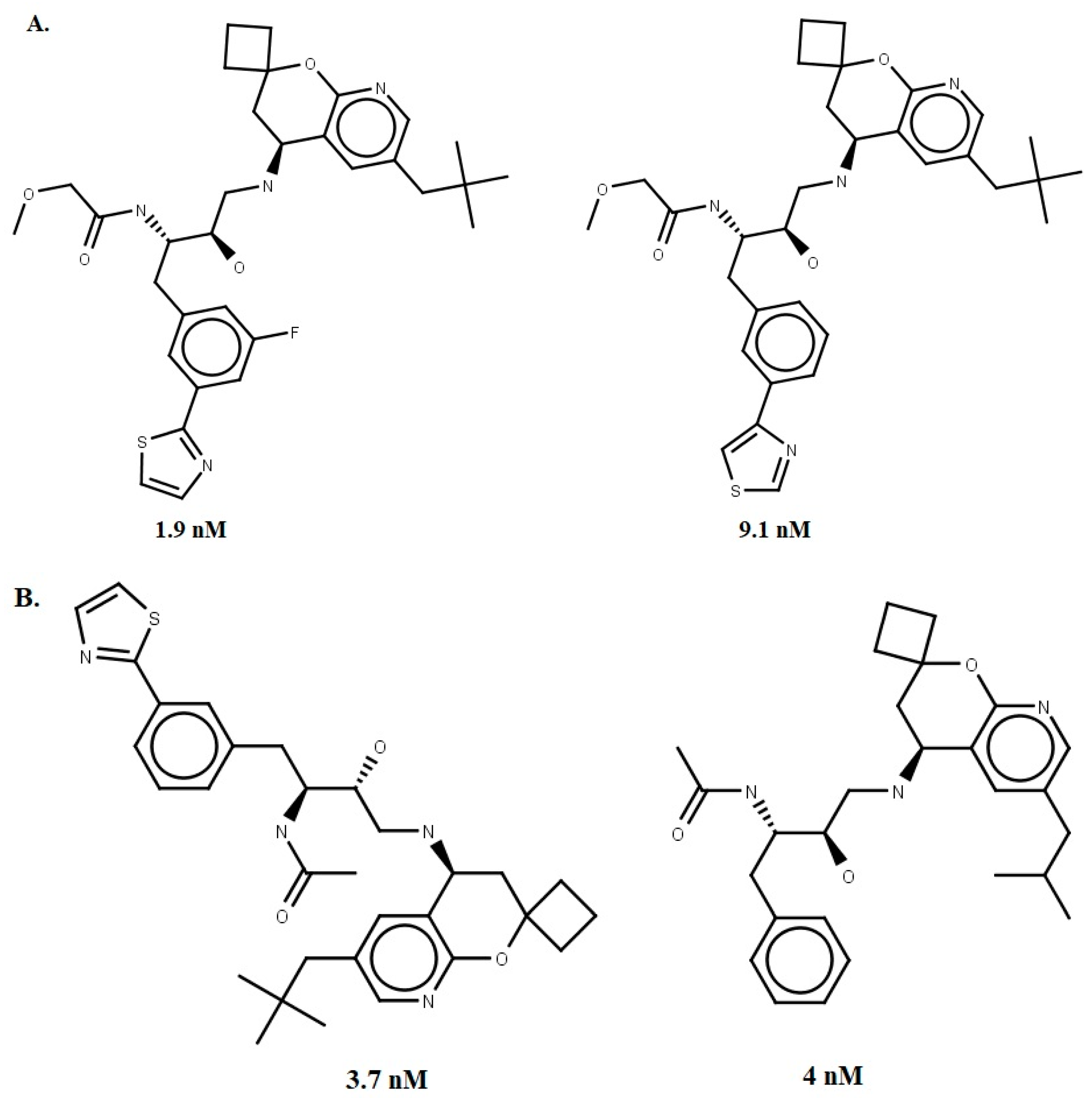
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J. Med. Chem. 2017, 61, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Kothandan, G.; Madhavan, T.; Gadhe, C.G.; Cho, S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res. 2013, 22, 1773–1787. [Google Scholar] [CrossRef]
- Leite, F.H.A. Planejamento e Avaliação de Novos Inibidores de Pteridina Redutase 1 (Ptr1) de Leishmania Major. Ph.D. Thesis, Universidade Estadual de Feira de Santana, Feira de Santana, Brazil, 2015. [Google Scholar]
- Bomfim, M.R.; Barbosa, D.B.; Carvalho, P.B.; Silva, A.M.; Oliveira, T.A.; Taranto, A.G.; Leite, F.H.A. Identification of potential human beta-secretase 1 inhibitors by hierarchical virtual screening and molecular dynamics. J. Biomol. Struct. Dyn. 2022, 41, 4560–4574. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the oral bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Degoey, D.A.; Chen, H.-J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar]
- Munoz-Torrero, D.; Camps, P. Dimeric and hybrid anti-Alzheimer drug candidates. Curr. Med. Chem. 2006, 13, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Türkeş, C.; Arslan, M.; Demir, Y.; Çoçaj, L.; Nixha, A.R.; Beydemir, Ş. Synthesis, biological evaluation and in silico studies of novel N-substituted phthalazine 103 sulfonamide compounds as potent carbonic anhydrase and acetylcholinesterase inhibitors. Bioorg. Chem. 2019, 89, 103004. [Google Scholar] [CrossRef]
- Senol, F.S.; Ślusarczyk, S.; Matkowski, A.; Pérez-Garrido, A.; Girón-Rodríguez, F.; Cerón-Carrasca, J.P.; Den-Haan, H.; Pena-García, J.; Pérez-Sánchez, H.; Domaradzki, K. Selective in vitro and in silico butyrylcholinesterase inhibitory activity of diterpenes and rosmarinic acid isolated from Perovskia atriplicifolia Benth. and Salvia glutinosa L. Phytochemistry 2016, 133, 33–44. [Google Scholar] [CrossRef]
- Wajid, S.; Khatoon, A.; Khan, M.A.; Zafar, H.; Kanwal, S.; Atta-Ur-Rahman; Choudhary, M.I.; Basha, F.Z. Microwave-assisted organic synthesis, structure-activity relationship, kinetics and molecular docking studies of non-cytotoxic benzamide derivatives as selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2019, 27, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Saha, A.; Roy, K. In silico modeling for dual inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes in Alzheimer’s disease. Comput. Biol. Chem. 2020, 88, 4030–4040. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Goyal, S.; Sharma, S.; Hamid, R.; Grover, A. Mechanist insights into mode of action of potent natural antagonists of BACE-1 for checking Alzheimer’s plaque pathology. Biochem. Biophys. Res. Commun. 2014, 443, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Semighini, E.P. In silico design of beta-secretase inhibitors in Alzheimer’s disease. Chem. Biol. Drug Des. 2014, 86, 284–290. [Google Scholar] [CrossRef]
- Dhanabalan, A.K.; Kesherwani, M.; Velmurugan, D.; Gunasekaran, K. Identification of new BACE1 inhibitors using pharmacophore and molecular dynamics simulations approach. J. Mol. Graph. Model. 2017, 76, 56–69. [Google Scholar] [CrossRef]
- Barbezan, A.B.; Martins, R.; Bueno, J.B.; Villavicencio, A.L.C.H. Ames test to detect mutagenicity of 2-alkylcyclobutanones: A review. J. Food Sci. 2017, 82, 1518–1522. [Google Scholar] [CrossRef]
- Modi, S.; Li, J.; Malcomber, S.; Moore, C.; Scott, A.; White, A.; Carmichael, P. Integrated in silico approaches for the prediction of Ames test mutagenicity. J. Comput. -Aided Mol. Des. 2012, 26, 1017–1033. [Google Scholar] [CrossRef]
- Vian, M.; Raitano, G.; Roncaglioni, A.; Benfenati, E. In silico model for mutagenicity (Ames test), taking into account metabolism. Mutagenesis 2019, 34, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Palakurti, R.; Vadrevu, R. Pharmacophore based 3D-QSAR modeling, virtual screening and docking for identification of potential inhibitors of B-secretase. Comput. Biol. Chem. 2017, 68, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chemaxon. Marvin Sketch, Version 15.4.20; Chemaxon: Budapest, Hungary, 2015.
- Tripos. SYBYL-X 2.0.; Tripos: St Louis, MO, USA, 2010; Volume 190. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. DUD Enhanced: Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Systat. SigmaPlot™, versão 12.0; A Scientific Data Management Company: Palo Alto, CA, USA, 2014.
- Manoharan, P.; Ghoshal, N. Fragment-based virtual screening approach and molecular dynamics simulation studies for identification of BACE1 inhibitor leads. J. Biomol. Struct. Dyn. 2017, 36, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P. Crystal structures of human cholinesterases in complex with Huprine W and Tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyrylcholinesterase. Biochem. J. 2013, 453, 3393–3399. [Google Scholar] [CrossRef]
- Winneroski, L.L.; Erickson, J.A.; Green, S.J.; Lopez, J.E.; Stout, S.L.; Porter, W.J.; Timm, D.E.; Audia, J.E.; Barberis, M.; Beck, J.P.; et al. Preparation and Biological Evaluation of BACE1 Inhibitors: Leveraging trans-Cyclopropyl Moieties as Ligand Efficient Conformational Constraints. Bioorg. Med. Chem. 2019, 28, 115–119. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J.; Vina, A. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Cole, J.C.; Nissink, J.W.M.; Taylor, R. Protein-ligand docking and virtual screening with GOLD. In Virtual Screening in Drug Discovery; Shoichet, B., Alvarez, J., Eds.; Taylor & Francis CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Energy (Kcal/mol) | Sterics | H_Bond | Mol_qry | Pareto |
|---|---|---|---|---|---|
| 01 | 21.74 | 756.00 | 155.60 | 9.09 | 00 |
| 02 | 42.36 | 746.60 | 151.50 | 15.59 | 00 |
| 03 | 20.65 | 687.30 | 157.00 | 14.42 | 00 |
| 04 | 50.89 | 713.40 | 161.20 | 8.47 | 00 |
| 05 | 1043.58 | 746.90 | 153.00 | 14.40 | 00 |
| 06 | 783.55 | 738.00 | 161.30 | 6.34 | 00 |
| 07 | 151,768.74 | 779.70 | 152.30 | 15.64 | 00 |
| 08 | 64.82 | 750.30 | 161.20 | 2.92 | 00 |
| 09 | 25.54 | 693.90 | 155.80 | 9.04 | 00 |
| 10 | 254.77 | 723.20 | 153.60 | 10.54 | 00 |
| Molecule | AChE * | BChE * | BACE-1 ** |
|---|---|---|---|
| ZINC45068352 | −9.6 | −10.4 | 43.32 |
| ZINC03873986 | −10.6 | −11.2 | 41.54 |
| ZINC71787288 | −9.6 | −10.6 | 41.91 |
| Molecule | MW (g/mol) | HBD | HBA | cLogP | RB | HBD + HBA | |
|---|---|---|---|---|---|---|---|
| ZINC45068352 | 486.58 | 0 | 7 | 4.77 | 89.45 | 5 | 7 |
| ZINC03873986 | 442.43 | 2 | 6 | 4.15 | 100.27 | 0 | 8 |
| ZINC71787288 | 444.44 | 0 | 7 | 5.38 | 61.53 | 3 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, G.O.; Araújo Neto, M.F.d.; Barbosa, D.B.; Bomfim, M.R.d.; Andrade, L.S.M.; Carvalho, P.B.d.; Oliveira, T.A.d.; Falkoski, D.L.; Maia, E.H.B.; Valle, M.S.; et al. Identification of Potential Multitarget Compounds against Alzheimer’s Disease through Pharmacophore-Based Virtual Screening. Pharmaceuticals 2023, 16, 1645. https://doi.org/10.3390/ph16121645
Mendes GO, Araújo Neto MFd, Barbosa DB, Bomfim MRd, Andrade LSM, Carvalho PBd, Oliveira TAd, Falkoski DL, Maia EHB, Valle MS, et al. Identification of Potential Multitarget Compounds against Alzheimer’s Disease through Pharmacophore-Based Virtual Screening. Pharmaceuticals. 2023; 16(12):1645. https://doi.org/10.3390/ph16121645
Chicago/Turabian StyleMendes, Géssica Oliveira, Moysés Fagundes de Araújo Neto, Deyse Brito Barbosa, Mayra Ramos do Bomfim, Lorena Silva Matos Andrade, Paulo Batista de Carvalho, Tiago Alves de Oliveira, Daniel Luciano Falkoski, Eduardo Habib Bechelane Maia, Marcelo Siqueira Valle, and et al. 2023. "Identification of Potential Multitarget Compounds against Alzheimer’s Disease through Pharmacophore-Based Virtual Screening" Pharmaceuticals 16, no. 12: 1645. https://doi.org/10.3390/ph16121645