
MPMABP: A CNN and Bi-LSTM-Based Method for Predicting Multi-Activities of Bioactive Peptides


Abstract
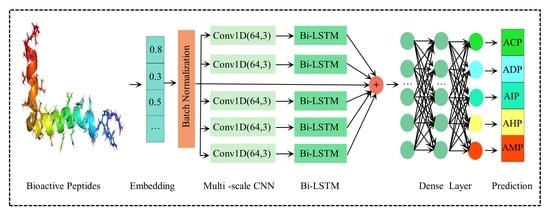
:
1. Introduction
2. Results and Discussion
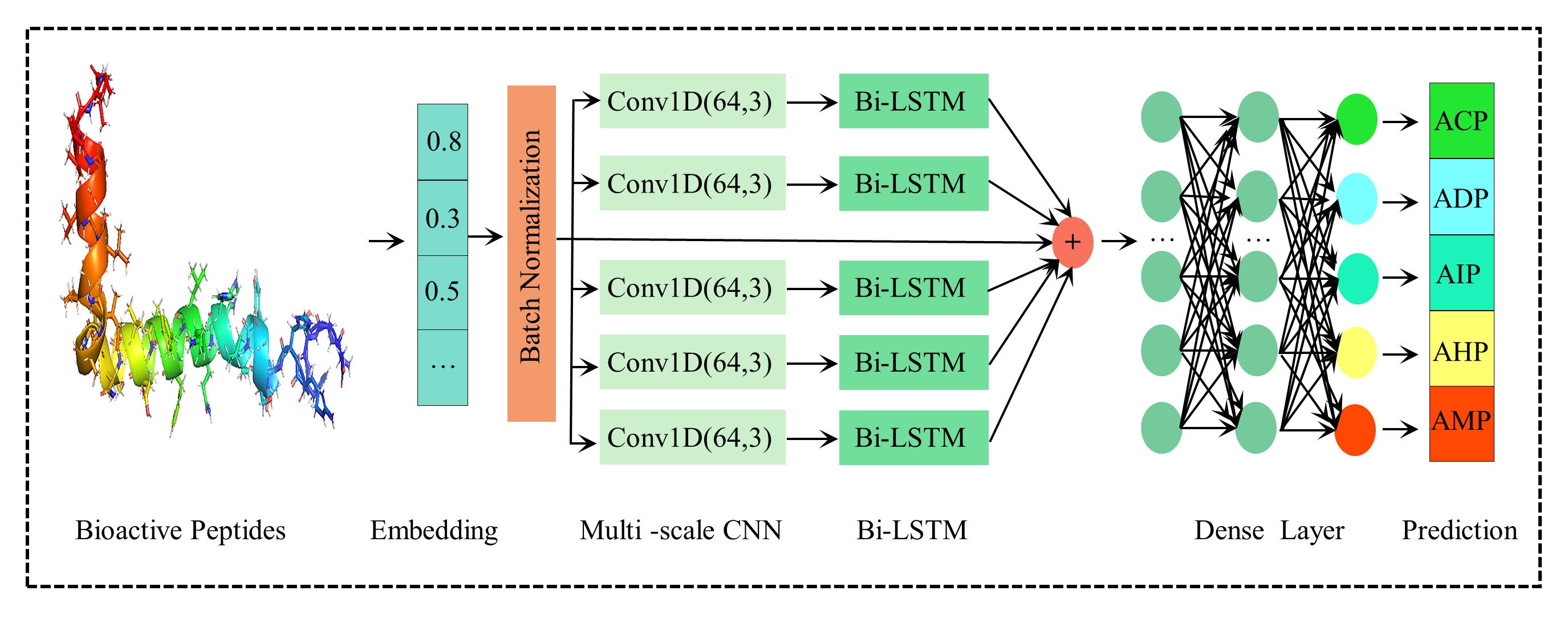
2.1. Optimization of Parameters
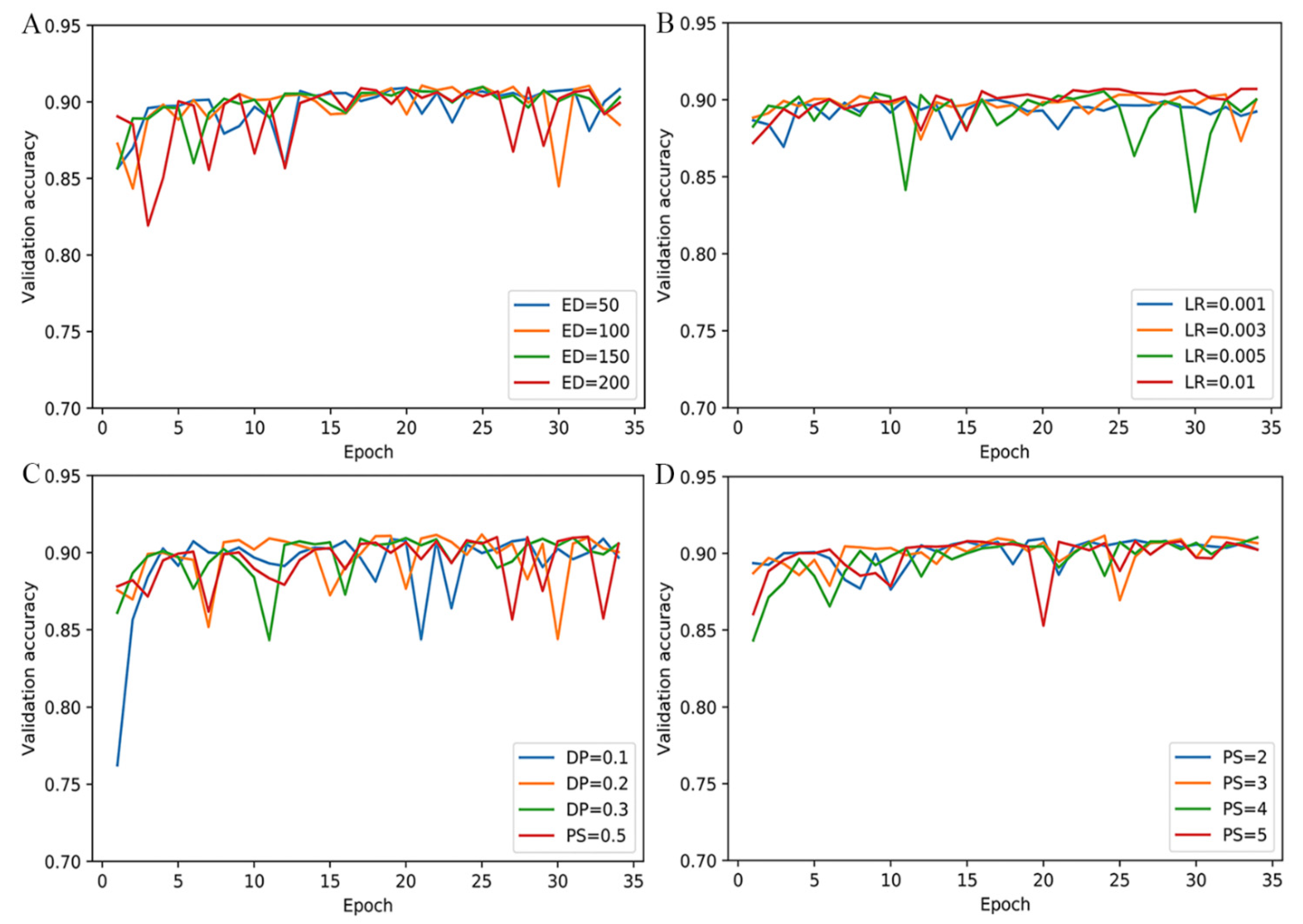
2.2. Comparison with State-of-the-Art Methods
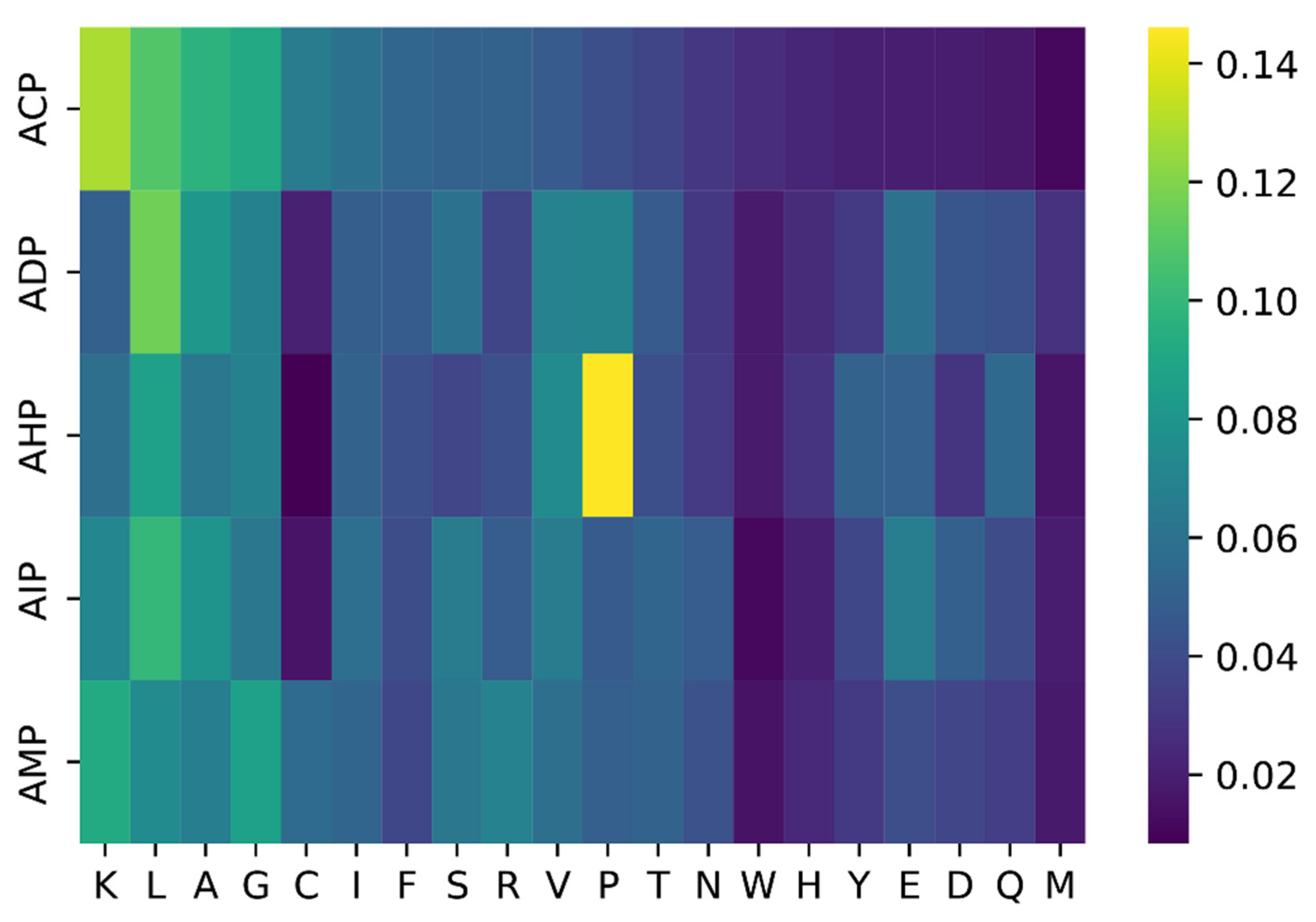
2.3. Case Study
2.4. Discussion
3. Materials and Methods
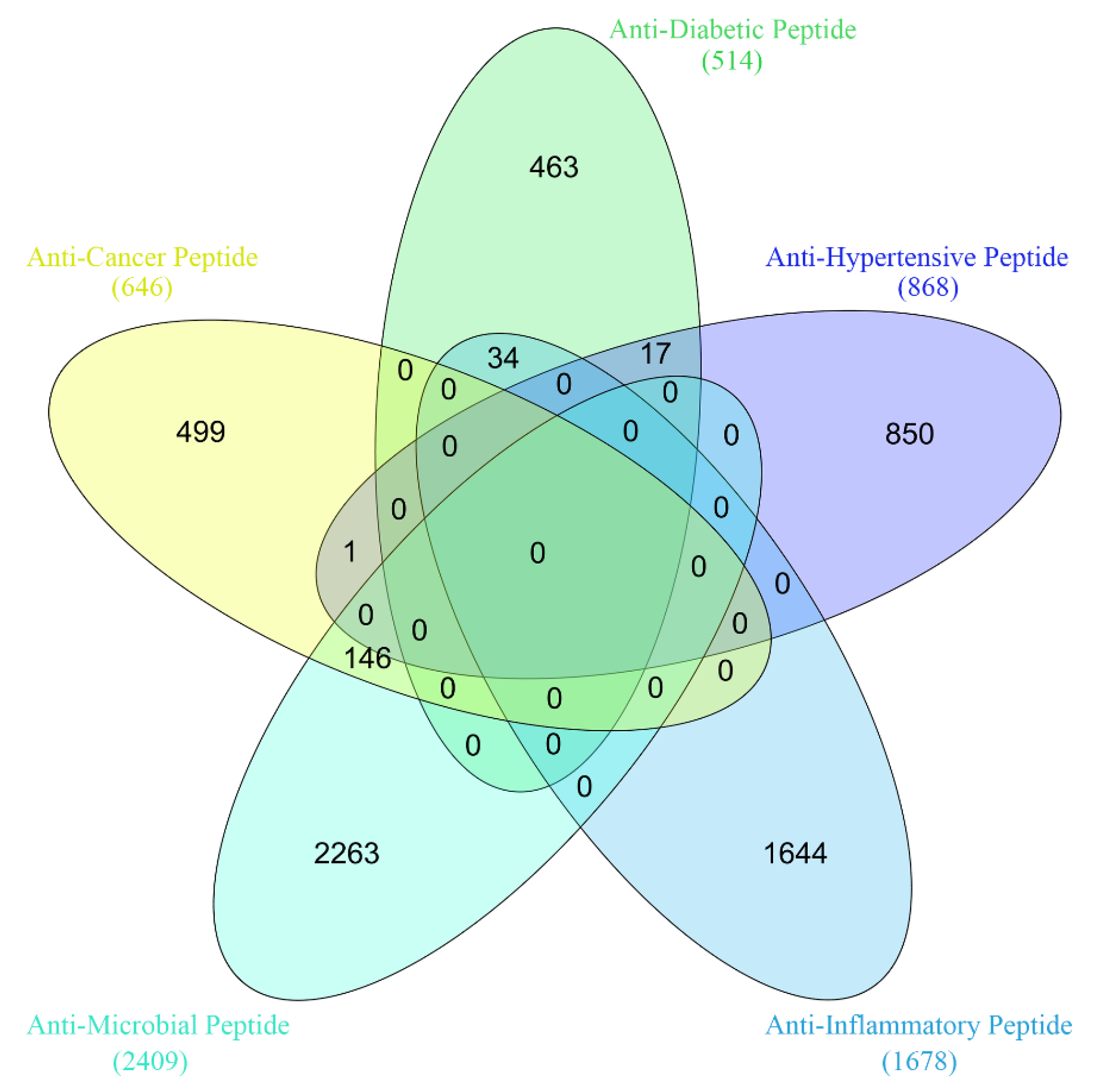
3.1. Datasets
3.2. Methodology
3.2.1. Embedding Layer
3.2.2. Multi-Scale CNN
3.2.3. Bi-LSTM
3.2.4. Pooling
3.3. ResNet
3.4. Fully Connected Layer
3.5. Validation and Evaluation Metrics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Luo, L.; Sun, X.; Ma, A. Bioactive Peptides: A Promising Alternative to Chemical Preservatives for Food Preservation. J. Agric. Food Chem. 2021, 69, 12369–12384. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, V.; Vasiljevic, T.; Donkor, O.N.; Mathai, M.L. A Review of Potential Marine-derived Hypotensive and Anti-obesity Peptides. Crit. Rev. Food Sci. Nutr. 2016, 56, 92–112. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Vázquez, A. Bioactive peptides: A review. Food Qual. Saf. 2017, 1, 29–46. [Google Scholar] [CrossRef]
- Kadam, S.U.; Tiwari, B.K.; Álvarez, C.; O’Donnell, C.P. Ultrasound applications for the extraction, identification and delivery of food proteins and bioactive peptides. Trends Food Sci. Technol. 2015, 46, 60–67. [Google Scholar] [CrossRef]
- Chalamaiah, M.; Yu, W.; Wu, J. Immunomodulatory and anticancer protein hydrolysates (peptides) from food proteins: A review. Food Chem. 2018, 245, 205–222. [Google Scholar] [CrossRef]
- Pavlicevic, M.; Marmiroli, N.; Maestri, E. Immunomodulatory peptides—A promising source for novel functional food production and drug discovery. Peptides 2022, 148, 170696. [Google Scholar] [CrossRef]
- Hussain, M.A.; Sumon, T.A.; Mazumder, S.K.; Ali, M.M.; Jang, W.J.; Abualreesh, M.H.; Sharifuzzaman, S.M.; Brown, C.L.; Lee, H.-T.; Lee, E.-W.; et al. Essential oils and chitosan as alternatives to chemical preservatives for fish and fisheries products: A review. Food Control 2021, 129, 108244. [Google Scholar] [CrossRef]
- Majumder, K.; Wu, J. Molecular Targets of Antihypertensive Peptides: Understanding the Mechanisms of Action Based on the Pathophysiology of Hypertension. Int. J. Mol. Sci. 2015, 16, 256–283. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Sharma, A.K.; Shastri, V.; Madhu, M.K.; Sharma, V.K. Prediction of anti-inflammatory proteins/peptides: An insilico approach. J. Transl. Med. 2017, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar]
- Zhao, J.; Bai, L.; Ren, X.-k.; Guo, J.; Xia, S.; Zhang, W.; Feng, Y. Co-immobilization of ACH11 antithrombotic peptide and CAG cell-adhesive peptide onto vascular grafts for improved hemocompatibility and endothelialization. Acta Biomater. 2019, 97, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Udenigwe, C.C. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci. Technol. 2014, 36, 137–143. [Google Scholar] [CrossRef]
- Li, Y.; Lyu, J.; Wu, Y.; Liu, Y.; Huang, G. PRIP: A Protein-RNA Interface Predictor Based on Semantics of Sequences. Life 2022, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Hussain, W.; Rasool, N.; Khan, Y.D. A sequence-based predictor of Zika virus proteins developed by integration of PseAAC and statistical moments. Comb. Chem. High Throughput Screen. 2020, 23, 797–804. [Google Scholar] [CrossRef]
- Aranha, M.P.; Spooner, C.; Demerdash, O.; Czejdo, B.; Smith, J.C.; Mitchell, J.C. Prediction of peptide binding to MHC using machine learning with sequence and structure-based feature sets. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129535. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M.; Peters, B.; Buus, S. Immunoinformatics: Predicting peptide–MHC binding. Annu. Rev. Biomed. Data Sci. 2020, 3, 191–215. [Google Scholar] [CrossRef]
- Yang, Z.; Yi, W.; Tao, J.; Liu, X.; Zhang, M.Q.; Chen, G.; Dai, Q. HPVMD-C: A disease-based mutation database of human papillomavirus in China. Database 2022, 2022, baac018. [Google Scholar] [CrossRef]
- Kong, R.; Xu, X.; Liu, X.; He, P.; Zhang, M.Q.; Dai, Q. 2SigFinder: The combined use of small-scale and large-scale statistical testing for genomic island detection from a single genome. BMC Bioinform. 2020, 21, 159. [Google Scholar] [CrossRef]
- Alzahrani, E.; Alghamdi, W.; Ullah, M.Z.; Khan, Y.D. Identification of stress response proteins through fusion of machine learning models and statistical paradigms. Sci. Rep. 2021, 11, 21767. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Y.; Chen, Y.; Dai, Q. MASQC: Next Generation Sequencing Assists Third Generation Sequencing for Quality Control in N6-Methyladenine DNA Identification. Front. Genet. 2020, 11, 269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Luo, W.; Lyu, J.; Yu, Z.-G.; Huang, G. CNNLSTMac4CPred: A Hybrid Model for N4-Acetylcytidine Prediction. Interdiscip. Sci. Comput. Life Sci. 2022, 14, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zheng, P.; Li, X.; Wu, H.; Wei, D.-Q.; Liu, Y.; Huang, G. Deep6mAPred: A CNN and Bi-LSTM-based deep learning method for predicting DNA N6-methyladenosine sites across plant species. Methods 2022, 204, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. iPhosS(Deep)-PseAAC: Identify Phosphoserine Sites in Proteins using Deep Learning on General Pseudo Amino Acid Compositions via Modified 5-Steps Rule. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. NPalmitoylDeep-PseAAC: A predictor of N-palmitoylation sites in proteins using deep representations of proteins and PseAAC via modified 5-steps rule. Curr. Bioinform. 2021, 16, 294–305. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. Sequence-based identification of arginine amidation sites in proteins using deep representations of proteins and PseAAC. Curr. Bioinform. 2020, 15, 937–948. [Google Scholar] [CrossRef]
- Shah, A.A.; Khan, Y.D. Identification of 4-carboxyglutamate residue sites based on position based statistical feature and multiple classification. Sci. Rep. 2020, 10, 16913. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. Optimization of serine phosphorylation prediction in proteins by comparing human engineered features and deep representations. Anal. Biochem. 2021, 615, 114069. [Google Scholar] [CrossRef]
- Huang, G.; Shen, Q.; Zhang, G.; Wang, P.; Yu, Z.G. LSTMCNNsucc: A Bidirectional LSTM and CNN-Based Deep Learning Method for Predicting Lysine Succinylation Sites. Biomed Res. Int. 2021, 2021, 9923112. [Google Scholar] [CrossRef]
- Onesime, M.; Yang, Z.; Dai, Q. Genomic Island Prediction via Chi-Square Test and Random Forest Algorithm. Comput. Math. Methods Med. 2021, 2021, 9969751. [Google Scholar] [CrossRef]
- Dai, Q.; Bao, C.; Hai, Y.; Ma, S.; Zhou, T.; Wang, C.; Wang, Y.; Huo, W.; Liu, X.; Yao, Y. MTGIpick allows robust identification of genomic islands from a single genome. Brief. Bioinform. 2018, 19, 361–373. [Google Scholar] [CrossRef]
- Roy, S.; Teron, R. BioDADPep: A Bioinformatics database for anti diabetic peptides. Bioinformation 2019, 15, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, C.; Chen, H.; Xue, J.; Guo, X.; Liang, M.; Chen, M. BioPepDB: An integrated data platform for food-derived bioactive peptides. Int. J. Food Sci. Nutr. 2018, 69, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P.S. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P.S. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Chaudhary, K.; Sharma, M.; Nagpal, G.; Chauhan, J.S.; Singh, S.; Gautam, A.; Raghava, G.P.S. AHTPDB: A comprehensive platform for analysis and presentation of antihypertensive peptides. Nucleic Acids Res. 2015, 43, D956–D962. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014, 2014, bau051. [Google Scholar] [CrossRef]
- Shtatland, T.; Guettler, D.; Kossodo, M.; Pivovarov, M.; Weissleder, R. PepBank—A database of peptides based on sequence text mining and public peptide data sources. BMC Bioinform. 2007, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, C.; Saavedra, Y.B.; Armijo-Galdames, B.; Amado-Hinojosa, J.; Olivera-Nappa, Á.; Sanchez-Daza, A.; Medina-Ortiz, D. Peptipedia: A user-friendly web application and a comprehensive database for peptide research supported by Machine Learning approach. Database 2021, 2021, baab055. [Google Scholar] [CrossRef]
- Khatun, M.S.; Hasan, M.M.; Kurata, H. PreAIP: Computational Prediction of Anti-inflammatory Peptides by Integrating Multiple Complementary Features. Front. Genet. 2019, 10, 129. [Google Scholar] [CrossRef]
- He, B.; Yang, S.; Long, J.; Chen, X.; Zhang, Q.; Gao, H.; Chen, H.; Huang, J. TUPDB: Target-Unrelated Peptide Data Bank. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 426–432. [Google Scholar] [CrossRef]
- Usmani, S.S.; Kumar, R.; Kumar, V.; Singh, S.; Raghava, G.P.S. AntiTbPdb: A knowledgebase of anti-tubercular peptides. Database 2018, 2018, bay025. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, D.Y.; Lee, G. Evolution of machine learning algorithms in the prediction and design of anticancer peptides. Curr. Protein Pept. Sci. 2020, 21, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, F.; Attique, M.; Khan, Y.D. AntiFlamPred: An Anti-Inflammatory Peptide Predictor for Drug Selection Strategies. CMC-Comput. Mater. Contin. 2021, 69, 1039–1055. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Chiangjong, W.; Hasan, M.M.; Nantasenamat, C.; Shoombuatong, W. Review and Comparative Analysis of Machine Learning-based Predictors for Predicting and Analyzing Anti-angiogenic Peptides. Curr. Med. Chem. 2022, 29, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Attique, M.; Farooq, M.S.; Khelifi, A.; Abid, A. Prediction of Therapeutic Peptides Using Machine Learning: Computational Models, Datasets, and Feature Encodings. IEEE Access 2020, 8, 148570–148594. [Google Scholar] [CrossRef]
- Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes 2021, 12, 137. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, J.; Zhao, L.; Zeng, X.; Liu, X. A novel antibacterial peptide recognition algorithm based on BERT. Brief. Bioinform. 2021, 22, bbab200. [Google Scholar] [CrossRef]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W.I. Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther.-Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef]
- Hussain, W. sAMP-PFPDeep: Improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks. Brief. Bioinform. 2022, 23, bbab487. [Google Scholar] [CrossRef]
- Arif, M.; Ahmed, S.; Ge, F.; Kabir, M.; Khan, Y.D.; Yu, D.-J.; Thafar, M. StackACPred: Prediction of anticancer peptides by integrating optimized multiple feature descriptors with stacked ensemble approach. Chemom. Intell. Lab. Syst. 2022, 220, 104458. [Google Scholar] [CrossRef]
- Hasan, M.M.; Schaduangrat, N.; Basith, S.; Lee, G.; Shoombuatong, W.; Manavalan, B. HLPpred-Fuse: Improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020, 36, 3350–3356. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.J.; Carper, D.L.; Spangler, M.K.; Carrell, A.A.; Rush, T.A.; Minter, S.J.; Weston, D.J.; Labbé, J.L. amPEPpy 1.0: A portable and accurate antimicrobial peptide prediction tool. Bioinformatics 2021, 37, 2058–2060. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. AtbPpred: A Robust Sequence-Based Prediction of Anti-Tubercular Peptides Using Extremely Randomized Trees. Comput. Struct. Biotechnol. J. 2019, 17, 972–981. [Google Scholar] [CrossRef]
- Usmani, S.S.; Bhalla, S.; Raghava, G.P.S. Prediction of Antitubercular Peptides from Sequence Information Using Ensemble Classifier and Hybrid Features. Front. Pharmacol. 2018, 9, 954. [Google Scholar] [CrossRef]
- Khatun, S.; Hasan, M.; Kurata, H. Efficient computational model for identification of antitubercular peptides by integrating amino acid patterns and properties. FEBS Lett. 2019, 593, 3029–3039. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.; Pu, L.; Tang, J.; Guo, F. AIEpred: An Ensemble Predictive Model of Classifier Chain to Identify Anti-Inflammatory Peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1831–1840. [Google Scholar] [CrossRef]
- Hasan, M.M.; Alam, M.A.; Shoombuatong, W.; Deng, H.-W.; Manavalan, B.; Kurata, H. NeuroPred-FRL: An interpretable prediction model for identifying neuropeptide using feature representation learning. Brief. Bioinform. 2021, 22, bbab167. [Google Scholar] [CrossRef]
- Ettayapuram Ramaprasad, A.S.; Singh, S.; Gajendra, P.S.R.; Venkatesan, S. AntiAngioPred: A server for prediction of anti-angiogenic peptides. PLoS ONE 2015, 10, e0136990. [Google Scholar] [CrossRef]
- Blanco, J.L.; Porto-Pazos, A.B.; Pazos, A.; Fernandez-Lozano, C. Prediction of high anti-angiogenic activity peptides in silico using a generalized linear model and feature selection. Sci. Rep. 2018, 8, 15688. [Google Scholar] [CrossRef] [Green Version]
- Khorsand, J.Z.; Ali Akbar, Y.; Kargar, M.; Ramin Shirali Hossein, Z.; Mahdevar, G. AntAngioCOOL: Computational detection of anti-angiogenic peptides. J. Transl. Med. 2019, 17, 71. [Google Scholar]
- Laengsri, V.; Nantasenamat, C.; Schaduangrat, N.; Nuchnoi, P.; Prachayasittikul, V.; Shoombuatong, W. TargetAntiAngio: A Sequence-Based Tool for the Prediction and Analysis of Anti-Angiogenic Peptides. Int. J. Mol. Sci. 2019, 20, 2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Wang, L.; Shi, L. AAPred-CNN: Accurate predictor based on deep convolution neural network for identification of anti-angiogenic peptides. Methods, 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. PIP-EL: A New Ensemble Learning Method for Improved Proinflammatory Peptide Predictions. Front. Immunol. 2018, 9, 1783. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Madhu, M.K.; Sharma, A.K.; Sharma, V.K. ProInflam: A webserver for the prediction of proinflammatory antigenicity of peptides and proteins. J. Transl. Med. 2016, 14, 178. [Google Scholar] [CrossRef] [Green Version]
- Khatun, M.S.; Hasan, M.M.; Shoombuatong, W.; Kurata, H. ProIn-Fuse: Improved and robust prediction of proinflammatory peptides by fusing of multiple feature representations. J. Comput.-Aided Mol. Des. 2020, 34, 1229–1236. [Google Scholar] [CrossRef]
- Chaudhary, K.; Kumar, R.; Singh, S.; Tuknait, A.; Gautam, A.; Mathur, D.; Anand, P.; Varshney, G.C.; Raghava, G.P.S. A Web Server and Mobile App for Computing Hemolytic Potency of Peptides. Sci. Rep. 2016, 6, 22843. [Google Scholar] [CrossRef]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.S.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Future Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application. Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P.S. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 2021, 22, bbaa153. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumar, S.; Ptv, L. ACPP: A Web Server for Prediction and Design of Anti-cancer Peptides. Int. J. Pept. Res. Ther. 2015, 21, 99–106. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Iqbal, M.; Jan, M.A. iACP-GAEnsC: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 2017, 79, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabir, M.; Arif, M.; Ahmad, S.; Ali, Z.; Swati, Z.N.K.; Yu, D.-J. Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom. Intell. Lab. Syst. 2018, 182, 158–165. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Rao, B.; Zhou, C.; Zhang, G.; Su, R.; Wei, L. ACPred-Fuse: Fusing multi-view information improves the prediction of anticancer peptides. Brief. Bioinform. 2020, 21, 1846–1855. [Google Scholar] [CrossRef]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther.-Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Charoenkwan, P.; Chiangjong, W.; Lee, V.S.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. Improved prediction and characterization of anticancer activities of peptides using a novel flexible scoring card method. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Su, R.; Zou, Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019, 35, 4272–4280. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Gao, R.; Zhang, Y.; De Marinis, Y. PTPD: Predicting therapeutic peptides by deep learning and word2vec. BMC Bioinform. 2019, 20, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.P.; Zou, Q. PPTPP: A novel therapeutic peptide prediction method using physicochemical property encoding and adaptive feature representation learning. Bioinformatics 2020, 36, 3982–3987. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yan, K.; Lv, H.; Liu, B. PreTP-EL: Prediction of therapeutic peptides based on ensemble learning. Brief. Bioinform. 2021, 22, bbab358. [Google Scholar] [CrossRef]
- He, W.; Jiang, Y.; Jin, J.; Li, Z.; Zhao, J.; Manavalan, B.; Su, R.; Gao, X.; Wei, L. Accelerating bioactive peptide discovery via mutual information-based meta-learning. Brief. Bioinform. 2022, 23, bbab499. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Tang, W.; Dai, R.; Yan, W.; Zhang, W.; Bin, Y.; Xia, E.; Xia, J. Identifying multi-functional bioactive peptide functions using multi-label deep learning. Brief. Bioinform. 2021, 23, bbab414. [Google Scholar] [CrossRef]
- Chung, J.; Gulcehre, C.; Cho, K.; Bengio, Y. Empirical evaluation of gated recurrent neural networks on sequence modeling. arXiv 2014, arXiv:1412.3555. [Google Scholar]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25, 1097–1105. [Google Scholar] [CrossRef]
- Kiperwasser, E.; Goldberg, Y. Simple and Accurate Dependency Parsing Using Bidirectional LSTM Feature Representations. Trans. Assoc. Comput. Linguist. 2016, 4, 313–327. [Google Scholar] [CrossRef]
- Fürnkranz, J.; Hüllermeier, E.; Mencía, E.L.; Brinker, K.J.M.l. Multilabel classification via calibrated label ranking. Mach. Learn. 2008, 73, 133–153. [Google Scholar] [CrossRef] [Green Version]
- Tsoumakas, G.; Vlahavas, I. Random k-labelsets: An ensemble method for multilabel classification. In Proceedings of the European Conference on Machine Learning, Warsaw, Poland, 17–21 September 2007; pp. 406–417. [Google Scholar]
- Wu, G.; Zheng, R.; Tian, Y.; Liu, D.J.N.N. Joint ranking SVM and binary relevance with robust low-rank learning for multi-label classification. Neural Netw. 2020, 122, 24–39. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wu, X.-Z.; Jiang, Y.; Zhou, Z.-H. Multi-label learning with deep forest. arXiv 2019, arXiv:1911.06557. [Google Scholar]
- Dong, G.; Zheng, L.; Huang, S.-H.; Gao, J.; Zuo, Y. Amino acid reduction can help to improve the identification of antimicrobial peptides and their functional activities. Front. Genet. 2021, 12, 549. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2019, 35, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, S. Accurate Prediction of Anti-hypertensive Peptides Based on Convolutional Neural Network and Gated Recurrent unit. Interdiscip. Sci. Comput. Life Sci. 2022, 1–6. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-based prediction of anti-inflammatory peptides using random forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef]
- Grønning, A.G.; Kacprowski, T.; Schéele, C.J.B.M. MultiPep: A hierarchical deep learning approach for multi-label classification of peptide bioactivities. Biol. Methods Protoc. 2021, 6, bpab021. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Fukushima, K.; Miyake, S. Neocognitron: A new algorithm for pattern recognition tolerant of deformations and shifts in position. Pattern Recognit. 1982, 15, 455–469. [Google Scholar] [CrossRef]
- Hubel, D.H.; Wiesel, T.N. Receptive fields, binocular interaction and functional architecture in the cat’s visual cortex. J. Physiol. 1962, 160, 106–154. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Boser, B.; Denker, J.; Henderson, D.; Howard, R.; Hubbard, W.; Jackel, L. Handwritten digit recognition with a back-propagation network. Adv. Neural Inf. Process. Syst. 1990, 2, 396–404. [Google Scholar]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going deeper with convolutions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 1–9. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Pearlmutter, B.A. Learning State Space Trajectories in Recurrent Neural Networks. Neural Comput. 1989, 1, 263–269. [Google Scholar] [CrossRef]
- Pearlmutter, B.A. Dynamic Recurrent Neural Networks. 1990. Unpublished work. Available online: https://mural.maynoothuniversity.ie/5505/ (accessed on 21 April 2022).
- Snyders, S.; Omlin, C.W. Inductive bias in recurrent neural networks. In Proceedings of the International Work-Conference on Artificial Neural Networks, Granada, Spain, 13–15 June 2001; pp. 339–346. [Google Scholar]
- Ullah, A.; Ahmad, J.; Muhammad, K.; Sajjad, M.; Baik, S.W. Action Recognition in Video Sequences using Deep Bi-Directional LSTM With CNN Features. IEEE Access 2018, 6, 1155–1166. [Google Scholar] [CrossRef]
- Siami-Namini, S.; Tavakoli, N.; Namin, A.S. The Performance of LSTM and BiLSTM in Forecasting Time Series. In Proceedings of the 2019 IEEE International Conference on Big Data (Big Data), Los Angeles, CA, USA, 9–12 December 2019; pp. 3285–3292. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | Super-Parameter | Value |
|---|---|---|
| Embedding | embedding dimensions | 100 |
| CNN layer 1 | number of kernels | 64 |
| size of kernels | 3 | |
| CNN layer 2 | number of kernels | 64 |
| size of kernels | 5 | |
| CNN layer 3 | number of kernels | 64 |
| size of kernels | 8 | |
| CNN layer 4 | number of kernels | 64 |
| size of kernels | 10 | |
| CNN layer 5 | number of kernels | 64 |
| size of kernels | 12 | |
| Pooling layer | size of pooling | 3 |
| stride | 1 | |
| Bi-LSTM layer | number of neurons | 32 |
| Dense1 | number of neurons | 64 |
| activation function | relu | |
| Dense2 | number of neurons | 128 |
| activation function | relu | |
| Dense3 | number of neurons | 5 |
| activation function | relu |
| Model | Precision | Coverage | Accuracy | Absolute True | Absolute False |
|---|---|---|---|---|---|
| MPMABP | 0.731 ± 0.011 | 0.738 ± 0.012 | 0.722 ± 0.010 | 0.696 ± 0.013 | 0.099 ± 0.006 |
| MLBP [87] | 0.697 ± 0.012 | 0.701 ± 0.014 | 0.695 ± 0.012 | 0.685 ± 0.011 | 0.109 ± 0.004 |
| Model | Precision | Coverage | Accuracy | Absolute True | Absolute False |
|---|---|---|---|---|---|
| MPMABP | 0.728 | 0.749 | 0.727 | 0.704 | 0.101 |
| MLBP [87] | 0.710 | 0.720 | 0.709 | 0.697 | 0.106 |
| CLR [92] | 0.667 | 0.677 | 0.666 | 0.655 | 0.133 |
| RAKEL [93] | 0.649 | 0.648 | 0.648 | 0.647 | 0.141 |
| MLDF [95] | 0.649 | 0.649 | 0.648 | 0.646 | 0.119 |
| RBRL [94] | 0.650 | 0.651 | 0.649 | 0.646 | 0.140 |
| Method | MPMABP | IAMP-RAAC [96] | mAHTPred [97] | AHPPred [98] | AIPpred [99] | |
|---|---|---|---|---|---|---|
| Type | ||||||
| AMP | 0.872 | 0.788 | - | - | - | |
| ACP | 0.505 | 0.333 | - | - | - | |
| AHP | 0.889 | - | 0.986 | 0.361 | - | |
| AIP | 0.914 | - | - | - | 0.827 | |
| Sequence | True labels | Prediction | ||
|---|---|---|---|---|
| MPMABP | MLBP [87] | MultiPep [100] | ||
| ACP-499 | ACP | ACP | ACP | AMP/anti-virus/ACP/anti-bacterial /anti-fungal |
| ADP-156 | ADP | ADP | ADP | ACE inhibitor/AHP |
| AHP-665 | AHP | AHP | AHP | Neuropeptide/peptidehormone |
| AIP-1046 | AIP | AIP | AIP | AMP/anti-bacterial |
| AMP-1389 | AMP | AMP | AMP | AMP/anti-bacterial |
| ACP-29 | ACP/AMP | ACP/AMP | AMP | ACP/anti-bacterial/anti-fungal |
| ACP-220 | ACP/AMP | ACP/AMP | None | AMP/anti-bacterial/anti-fungal |
| ADP-463 | ADP/AHP | ADP/AHP | ADP | ADP |
| AIP-1050 | AIP/ADP | ADP/AIP | ADP/AHP | ADP |
| AHP-483 | AHP/ACP | AHP | AHP | Antioxidative/ACE inhibitor/AHP |
| Model | Precision | Coverage | Accuracy | Absolute True | Absolute False |
|---|---|---|---|---|---|
| MPMABPwr a | 0.702 | 0.723 | 0.701 | 0.678 | 0.108 |
| MPMABPwr b | 0.697 ± 0.013 | 0.704 ± 0.022 | 0.688 ± 0.013 | 0.663 ± 0.013 | 0.105 ± 0.003 |
| MPMABPsc a | 0.697 | 0.719 | 0.696 | 0.672 | 0.109 |
| MPMABPsc b | 0.704 ± 0.019 | 0.710 ±0.023 | 0.694 ± 0.019 | 0.668 ± 0.018 | 0.103 ± 0.006 |
| Model | Precision | Coverage | Accuracy | Absolute True | Absolute False |
|---|---|---|---|---|---|
| MPMABP | 0.731 ± 0.011 | 0.738 ± 0.012 | 0.722 ± 0.010 | 0.696 ± 0.013 | 0.099 ± 0.006 |
| No CNN | 0.724 ± 0.011 | 0.729 ± 0.010 | 0.714 ± 0.011 | 0.689 ± 0.013 | 0.101 ± 0.004 |
| No LSTM | 0.708 ± 0.017 | 0.708 ± 0.014 | 0.698 ± 0.017 | 0.678 ± 0.020 | 0.102 ± 0.004 |
| Degeneration | 0.725 ± 0.015 | 0.733 ± 0.015 | 0.716 ± 0.014 | 0.688 ± 0.013 | 0.101 ± 0.009 |
| Model | Precision | Coverage | Accuracy | Absolute True | Absolute False |
|---|---|---|---|---|---|
| MPMABP | 0.728 | 0.749 | 0.727 | 0.704 | 0.101 |
| No CNN | 0.676 | 0.688 | 0.675 | 0.662 | 0.105 |
| No LSTM | 0.659 | 0.670 | 0.658 | 0.645 | 0.109 |
| Degeneration | 0.690 | 0.708 | 0.689 | 0.670 | 0.111 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, X.; Liu, Y.; Yao, Y.; Huang, G. MPMABP: A CNN and Bi-LSTM-Based Method for Predicting Multi-Activities of Bioactive Peptides. Pharmaceuticals 2022, 15, 707. https://doi.org/10.3390/ph15060707
Li Y, Li X, Liu Y, Yao Y, Huang G. MPMABP: A CNN and Bi-LSTM-Based Method for Predicting Multi-Activities of Bioactive Peptides. Pharmaceuticals. 2022; 15(6):707. https://doi.org/10.3390/ph15060707
Chicago/Turabian StyleLi, You, Xueyong Li, Yuewu Liu, Yuhua Yao, and Guohua Huang. 2022. "MPMABP: A CNN and Bi-LSTM-Based Method for Predicting Multi-Activities of Bioactive Peptides" Pharmaceuticals 15, no. 6: 707. https://doi.org/10.3390/ph15060707