
The Glycosylation of Immune Checkpoints and Their Applications in Oncology

Abstract
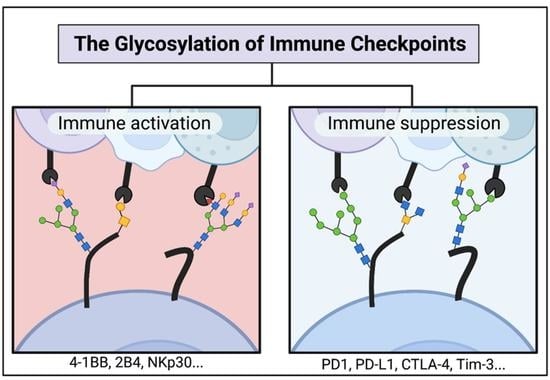
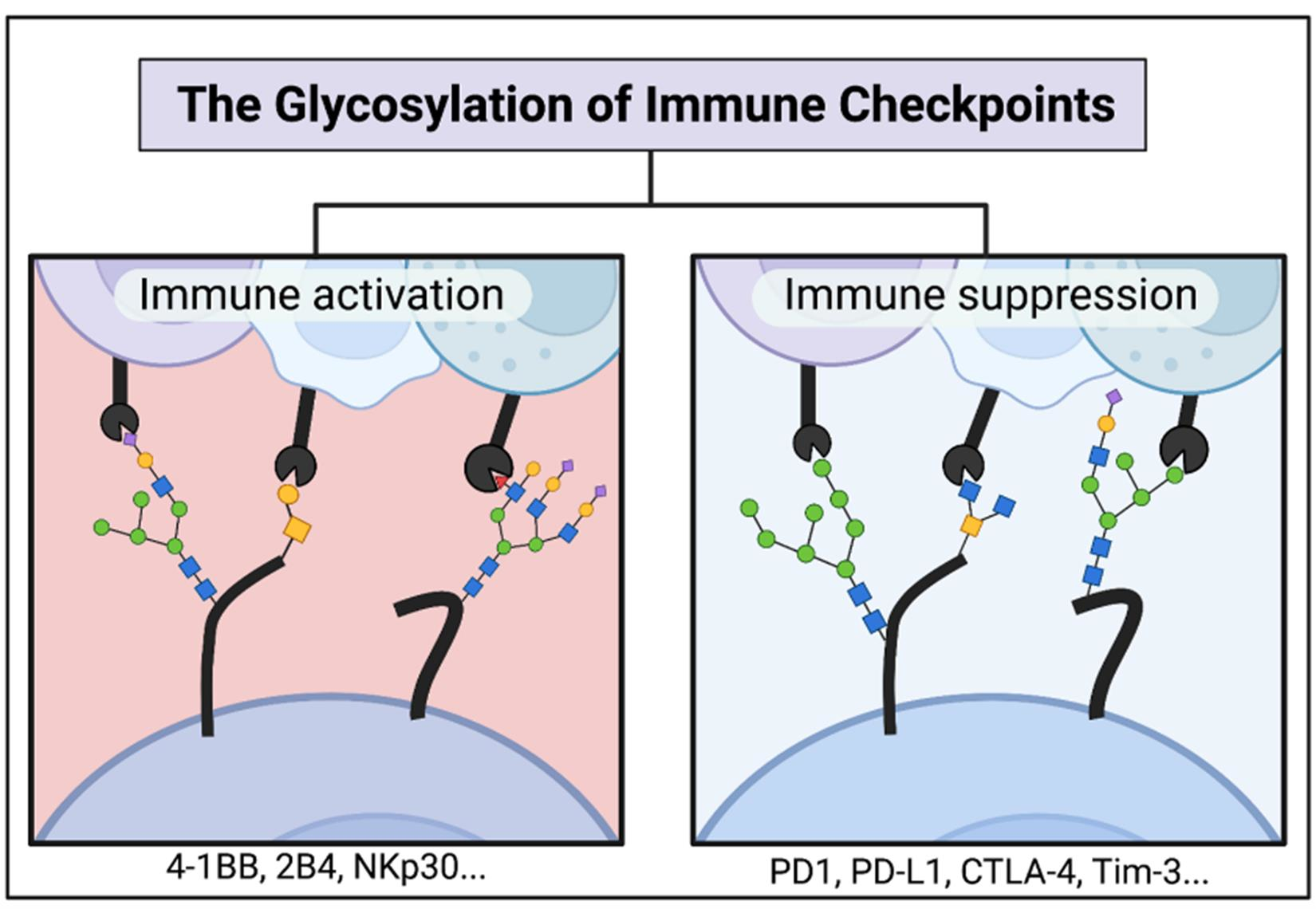
:
1. Introduction
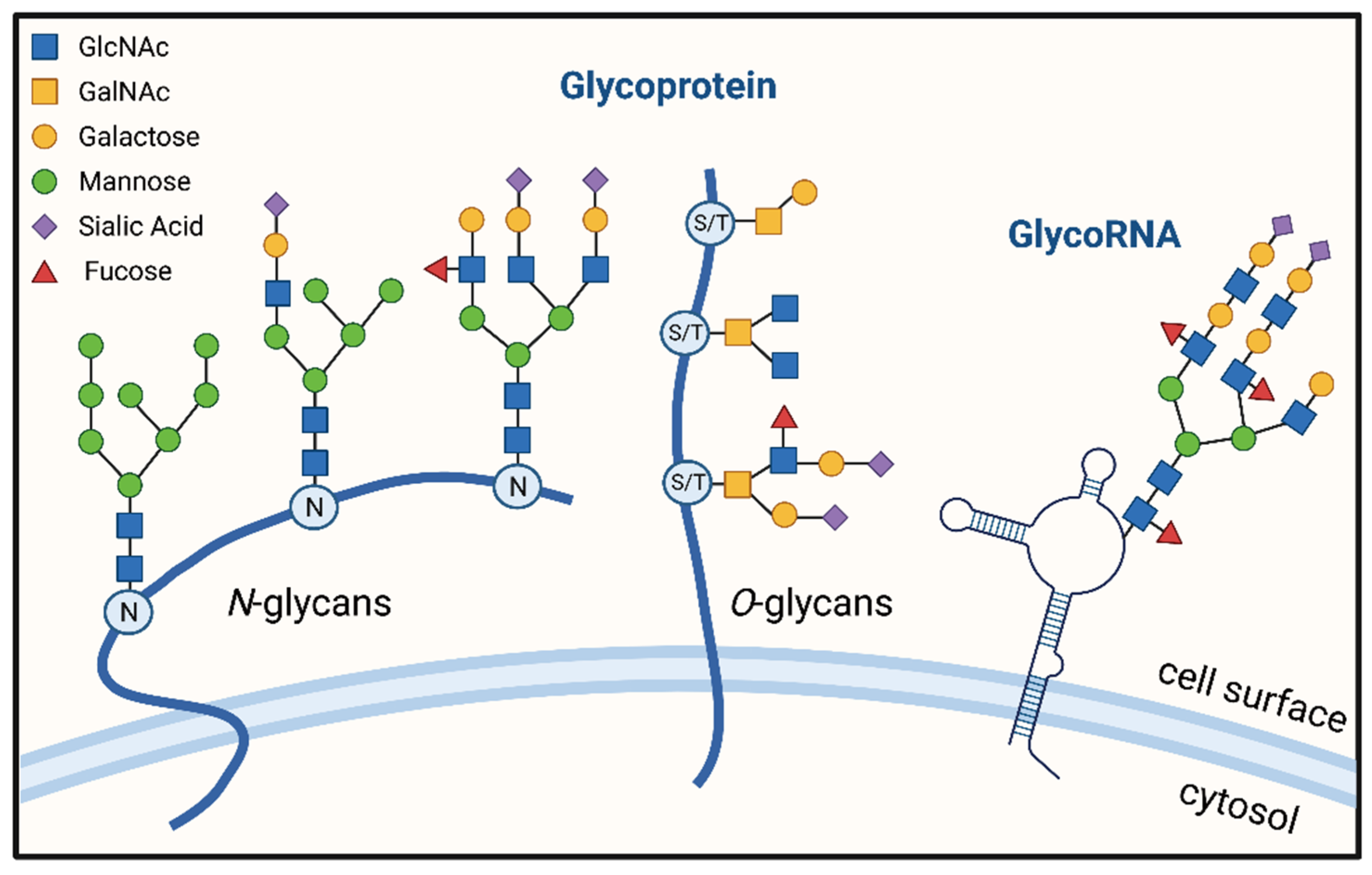
2. The Process of Glycosylation of Proteins
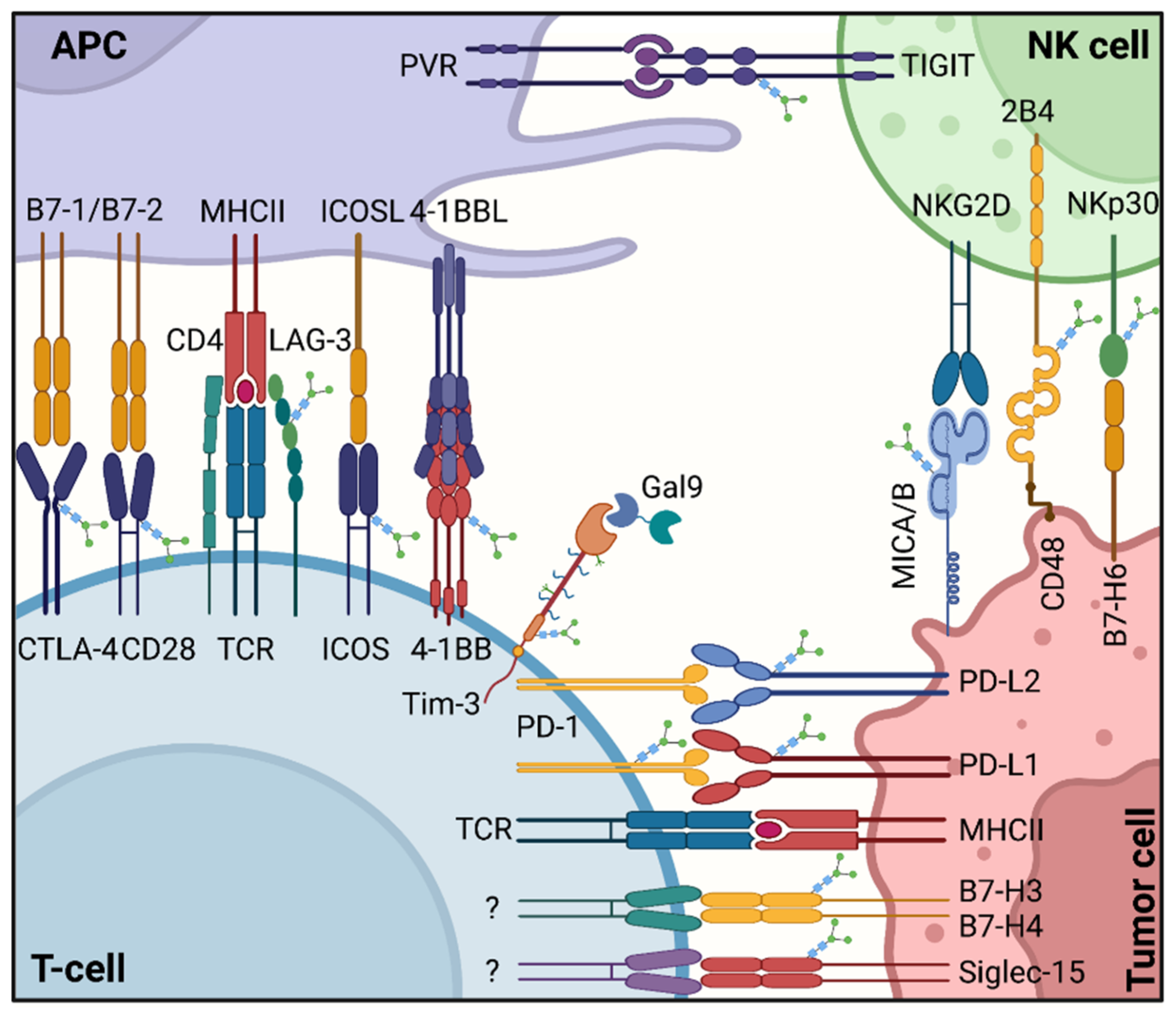
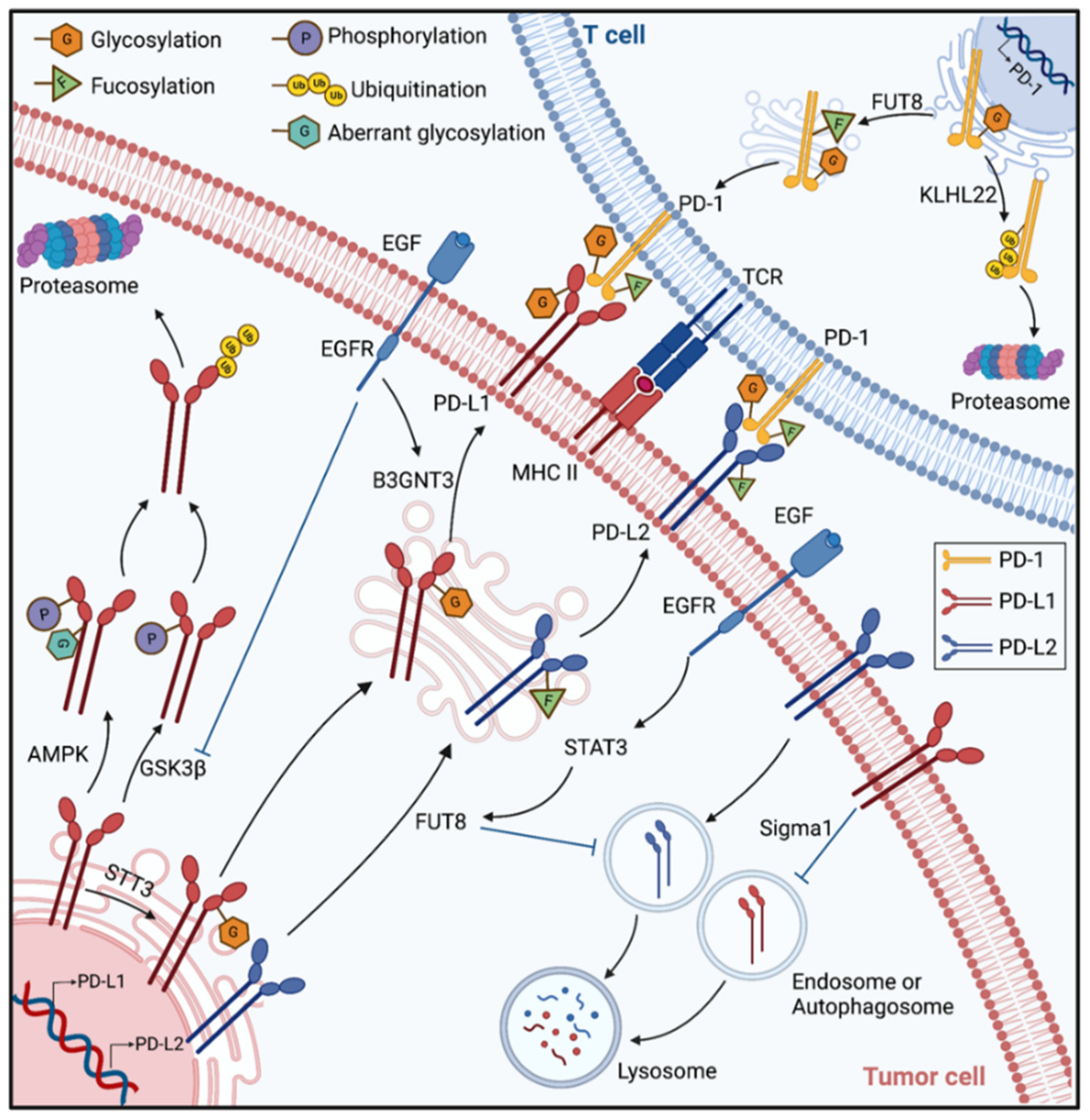
3. N-Glycosylated Immune Checkpoints
3.1. B7-CD28 Superfamily
3.2. Immune Checkpoints Outside the B7 Superfamily
4. O-Glycosylation
5. GlycoRNA
6. Applications in Glycosylation-Targeted Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Drug Name | Drug Type | Target | Study Phase |
|---|---|---|---|---|
| [44] | STM408 | Monoclonal antibody | N58-glycosylated PD-1 | Preclinical study |
| [108] | MW11-h317 | Monoclonal antibody | N58-glycosylated PD-1 | Preclinical study |
| [109] | mAb059c | Monoclonal antibody | N58-glycosylated PD-1 | Preclinical study |
| [50] | STM108 | Monoclonal antibody | N-glycosylated PD-L1 | Preclinical study |
| [111] | BMS1166 | Small molecule | N-glycosylated PD-L1 | Preclinical study |
| [43,44,66] | 2F-Fuc | Glucose analog | FUT8 | Preclinical study |
| [67,112,113,114] | NGI-1 | Small molecule | STT3A/B | Preclinical study |
| [98,115,116] | 2-DG | Glucose analog | Hexokinase | Preclinical study |
| [119] | SEA-TGT | Monoclonal antibody | TIGIT | Clinical trial |
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD-1 | programmed cell death protein-1 |
| PD-L1/2 | programmed death-ligand 1/2 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| TIM-3 | T-cell immunoglobulin and mucin domain-3 |
| LAG-3 | lymphocyte activation gene-3 |
| TIGIT | T cell immunoreceptor with immunoglobulin and ITIM domains |
| PTMs | Post-translational Modifications |
| GPI | glycosylphosphatidylinositol |
| ER | endoplasmic reticulum |
| OST | oligosaccharyltransferase |
| ERAD | ER-associated degradation |
| ERLAD | ER-to-lysosomes-associated degradation |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| HBP | hexosamine biosynthetic pathway |
| OGT | O-GlcNAc transferase |
| OGA | O-GlcNAcase |
| TCR | T-cell-receptor |
| MHC | major histocompatibility complex |
| APCs | antigen-presenting cells |
| FUT8 | Fucosyltransferase 8 |
| B3GNT3 | β-1,3-N-acetylglucosaminyl-transferase |
| CSCs | Cancer stem cells |
| EMT | epithelial-mesenchymal transition |
| GSK3β | glycogen synthase kinase 3β |
| TNBC | Triple-Negative Breast Cancer |
| AMPK | Adenosine 5′-monophosphate (AMP)-activated protein kinase |
| CTL | cytotoxic T lymphocyte |
| IHC | immunohistochemical |
| FFPE | Formalin-fixed paraffin-embedding |
| PNGase | F peptide-N-glycosidase F; |
| HNSCC | head and neck squamous cell carcinoma |
| EGF/EGFR | Epidermal Growth Factor/Epidermal Growth Factor Receptor |
| ICOS/ICOSL | inducible co-stimulator/ inducible co-stimulator ligand |
| NK cells | natural killer cells |
| NCRs | natural cytotoxicity receptors |
| LSECtin | liver sinusoidal endothelial cell lectin |
| TIGIT | T cell immunoreceptor with immunoglobulin and ITIM domains |
| CRD | cysteine-rich domains |
| Gal-9 | galectin-9 |
| Siglec | Sialic acid-binding immunoglobulin-like lectin |
| IAP | integrin-associated protein |
| SIRPα | signal regulatory protein α |
| TAMs | tumor-associated macrophages |
| NKG2D | natural killer group 2D |
| MICA/B | MHC class I polypeptide-related sequence A/B |
| SLAM | Signaling Lymphocyte Activation Molecule |
| DC | dendritic cells |
| SRPK2 | serine/arginine-rich protein kinase 2 |
| PGK1 | phosphoglycerate kinase 1 |
| TCA | tricarboxylic acid |
| 2F-Fuc | 2-fluoro-L-Fucose |
| 2-DG | 2-deoxy-glucose |
| PARP | Poly ADP-ribose polymerase |
| Fab | fragment antigen binding |
| Fc | fragment crystallizable |
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
References
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, I.; Bazhin, A.V. Metabolic Checkpoints: Novel Avenues for Immunotherapy of Cancer. Front. Immunol. 2018, 9, 1816. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrao, P.; Bork, P.; Krogan, N.J.; Van Noort, V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013, 9, 714. [Google Scholar] [CrossRef]
- Tokarz, P.; Woźniak, K. SENP Proteases as Potential Targets for Cancer Therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef]
- Wu, S.; Xia, Y.; Liu, X.; Sun, J. Vitamin D receptor deletion leads to reduced level of IκBα protein through protein translation, protein–protein interaction, and post-translational modification. Int. J. Biochem. Cell Biol. 2010, 42, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, C.; Jiang, H.; Yang, P.; Lu, H. Fishing the PTM proteome with chemical approaches using functional solid phases. Chem. Soc. Rev. 2015, 44, 8260–8287. [Google Scholar] [CrossRef]
- Huang, H.; Arighi, C.; Ross, K.E.; Ren, J.; Julie, C.; Chen, S.-C.; Wang, Q.; Cowart, J.; Vijay-Shanker, K.; Wu, C.H. iPTMnet: An integrated resource for protein post-translational modification network discovery. Nucleic Acids Res. 2017, 46, D542–D550. [Google Scholar] [CrossRef]
- Li, W.; Li, F.; Zhang, X.; Lin, H.-K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target. Ther. 2021, 6, 422. [Google Scholar] [CrossRef]
- Ferreira, J.A.; Magalhães, A.; Gomes, J.; Peixoto, A.; Gaiteiro, C.; Fernandes, E.; Santos, L.L.; Reis, C.A. Protein glycosylation in gastric and colorectal cancers: Toward cancer detection and targeted therapeutics. Cancer Lett. 2017, 387, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell 2021, 184, 3109–3124. [Google Scholar] [CrossRef]
- Silsirivanit, A. Glycosylation markers in cancer. Adv. Clin. Chem. 2019, 89, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein Glycosylation in Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 473–510. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2017, 9, 1380–1402. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Kim, A.; Lim, S.-O. Glycosylation of Immune Receptors in Cancer. Cells 2021, 10, 1100. [Google Scholar] [CrossRef]
- Bhargava, C.; Dürkop, H.; Zhao, X.; Weng, A.; Melzig, M.F.; Fuchs, H. Targeted dianthin is a powerful toxin to treat pancreatic carcinoma when applied in combination with the glycosylated triterpene SO1861. Mol. Oncol. 2017, 11, 1527–1543. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, J.; Lubman, D.M.; Gao, C. Aberrant glycosylation and cancer biomarker discovery: A promising and thorny journey. Clin. Chem. Lab. Med. 2019, 57, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lafite, P.; Daniellou, R. Rare and unusual glycosylation of peptides and proteins. Nat. Prod. Rep. 2012, 29, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P. Golgi Glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and Posttranslational N-Glycosylation of Polypeptides by Distinct Mammalian OST Isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Mammadova-Bach, E.; Jaeken, J.; Gudermann, T.; Braun, A. Platelets and Defective N-Glycosylation. Int. J. Mol. Sci. 2020, 21, 5630. [Google Scholar] [CrossRef] [PubMed]
- Dobrica, M.-O.; Lazar, C.; Branza-Nichita, N. N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis. Cells 2020, 9, 1404. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Fregno, I.; Fasana, E.; Soldà, T.; Galli, C.; Molinari, M. N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation. EMBO J. 2021, 40, e107240. [Google Scholar] [CrossRef] [PubMed]
- Özcan, S.; Andrali, S.S.; Cantrell, J.E.L. Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 2010, 1799, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Ramirez, D.H.; Yang, B.; D’Souza, A.K.; Aonbangkhen, C.; Wong, S.; Woo, C.M. Target protein deglycosylation in living cells by a nanobody-fused split O-GlcNAcase. Nat. Chem. Biol. 2021, 17, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Cao, M.; Ruan, X.; Jiang, L.; Lee, S.; Lemanek, A.; Ghassemian, M.; Pizzo, D.P.; Wan, Y.; Qiao, Y.; et al. Cancer-cell-secreted miR-122 suppresses O-GlcNAcylation to promote skeletal muscle proteolysis. Nat. Cell Biol. 2022, 24, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gupta, V.K.; Dauer, P.; Kesh, K.; Hadad, R.; Giri, B.; Chandra, A.; Dudeja, V.; Slawson, C.; Banerjee, S.; et al. O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics 2019, 9, 3410–3424. [Google Scholar] [CrossRef] [PubMed]
- Masbuchin, A.N.; Rohman, M.S.; Liu, P.-Y. Role of Glycosylation in Vascular Calcification. Int. J. Mol. Sci. 2021, 22, 9829. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7–CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Bretscher, P.A. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.Y.; Mikolajczak, S.; Yoshida, T.; Yoshida, R.; Kelvin, D.J.; Ochi, A. CD28 T cell costimulatory receptor function is negatively regulated by N-linked carbohydrates. Biochem. Biophys. Res. Commun. 2004, 317, 60–67. [Google Scholar] [CrossRef]
- Alegre, M.-L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef]
- Littman, D.R. Releasing the Brakes on Cancer Immunotherapy. Cell 2015, 162, 1186–1190. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Chikuma, S.; Kondo, T.; Hibino, S.; Machiyama, H.; Yokosuka, T.; Nakano, M.; Yoshimura, A. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. 2017, 20, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, C.-W.; Chung, E.M.; Yang, R.; Kim, Y.-S.; Park, A.H.; Lai, Y.-J.; Yang, Y.; Wang, Y.-H.; Liu, J.; et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020, 80, 2298–2310. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Clarke, M.F. Self-renewal and solid tumor stem cells. Oncogene 2004, 23, 7274–7282. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Liang, M.; Lai, M.; Shang, L.; Deng, X.; Su, X. KYA1797K down-regulates PD-L1 in colon cancer stem cells to block immune evasion by suppressing the β-catenin/STT3 signaling pathway. Int. Immunopharmacol. 2020, 78, 106003. [Google Scholar] [CrossRef]
- Zhou, X.A.; Zhou, J.; Zhao, L.; Yu, G.; Zhan, J.; Shi, C.; Yuan, R.; Wang, Y.; Chen, C.; Zhang, W.; et al. KLHL22 maintains PD-1 homeostasis and prevents excessive T cell suppression. Proc. Natl. Acad. Sci. USA 2020, 117, 28239–28250. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-H.; Wang, Y.-N.; Xia, W.; Chen, C.-H.; Rau, K.-M.; Ye, L.; Wei, Y.; Chou, C.-K.; Wang, S.-C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Okadome, K.; Baba, Y.; Nomoto, D.; Yagi, T.; Kalikawe, R.; Harada, K.; Hiyoshi, Y.; Nagai, Y.; Ishimoto, T.; Iwatsuki, M.; et al. Prognostic and clinical impact of PD-L2 and PD-L1 expression in a cohort of 437 oesophageal cancers. Br. J. Cancer 2020, 122, 1535–1543. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, Z.; Hu, R.; Wang, Y.; Wang, Y.; Su, Z.; Zhang, X.; Yang, J.; Mei, M.; Ren, Y.; et al. PD-L2 glycosylation promotes immune evasion and predicts anti-EGFR efficacy. J. Immunother. Cancer 2021, 9, e002699. [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Fukui, R.; Suzuki, Y.; Kajihara, Y.; Kinoshita, M.; Kakehi, K.; Hojo, H.; Tezuka, K.; Tsuji, T. Definitive evidence that a single N-glycan among three glycans on inducible costimulator is required for proper protein trafficking and ligand binding. Biochem. Biophys. Res. Commun. 2010, 391, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Rujas, E.; Cui, H.; Sicard, T.; Semesi, A.; Julien, J.-P. Structural characterization of the ICOS/ICOS-L immune complex reveals high molecular mimicry by therapeutic antibodies. Nat. Commun. 2020, 11, 5066. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, M.; Shah, U.A.; Liu, W.; Zhao, A.; Schoenberg, M.P.; Zang, X. The third group of the B7- CD 28 immune checkpoint family: HHLA 2, TMIGD 2, B7x, and B7-H3. Immunol. Rev. 2017, 276, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.-S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.-H.; Wong, C.-H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zhang, H.-L.; Li, Z.-L.; Du, T.; Chen, Y.-H.; Wang, Y.; Ni, H.-H.; Zhang, K.-M.; Mai, J.; Hu, B.-X.; et al. FUT8-mediated aberrant N-glycosylation of B7H3 suppresses the immune response in triple-negative breast cancer. Nat. Commun. 2021, 12, 2672. [Google Scholar] [CrossRef]
- Song, X.; Zhou, Z.; Li, H.; Xue, Y.; Lu, X.; Bahar, I.; Kepp, O.; Hung, M.-C.; Kroemer, G.; Wan, Y. Pharmacologic Suppression of B7-H4 Glycosylation Restores Antitumor Immunity in Immune-Cold Breast Cancers. Cancer Discov. 2020, 10, 1872–1893. [Google Scholar] [CrossRef]
- Hartmann, J.; Tran, T.-V.; Kaudeer, J.; Oberle, K.; Herrmann, J.; Quagliano, I.; Abel, T.; Cohnen, A.; Gatterdam, V.; Jacobs, A.; et al. The Stalk Domain and the Glycosylation Status of the Activating Natural Killer Cell Receptor NKp30 Are Important for Ligand Binding. J. Biol. Chem. 2012, 287, 31527–31539. [Google Scholar] [CrossRef]
- Skořepa, O.; Pazicky, S.; Kalousková, B.; Bláha, J.; Abreu, C.; Ječmen, T.; Rosůlek, M.; Fish, A.; Sedivy, A.; Harlos, K.; et al. Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers 2020, 12, 1998. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, B.J.; Mason, G.H.; Greenshields-Watson, A.; Triebel, F.; Gallimore, A.; Cole, D.K.; Godkin, A. Molecular characterization of HLA class II binding to the LAG-3 T cell co-inhibitory receptor. Eur. J. Immunol. 2020, 51, 331–341. [Google Scholar] [CrossRef]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-cell Responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Lambert, L.J.; Ely, Z.A.; Pattada, N.B.; Bhutkar, A.; Eng, G.; Mercer, K.L.; Garcia, A.P.; Lin, L.; Rideout, W.M.; et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 2021, 39, 1342–1360. [Google Scholar] [CrossRef]
- Lin, Y.-X.; Hung, M.-C.; Hsu, J.-L.; Hsu, J.-M. The N-linked glycosylations of TIGIT Asn32 and Asn101 facilitate PVR/TIGIT interaction. Biochem. Biophys. Res. Commun. 2021, 562, 9–14. [Google Scholar] [CrossRef]
- Vinay, D.S.; Kwon, B.S. 4-1BB signaling beyond T cells. Cell. Mol. Immunol. 2011, 8, 281–284. [Google Scholar] [CrossRef]
- Li, Y.; Tan, S.; Zhang, C.; Chai, Y.; He, M.; Zhang, C.W.-H.; Wang, Q.; Tong, Z.; Liu, K.; Lei, Y.; et al. Limited Cross-Linking of 4-1BB by 4-1BB Ligand and the Agonist Monoclonal Antibody Utomilumab. Cell Rep. 2018, 25, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitra, A.; Doukov, T.; Croft, M.; Zajonc, D.M. Crystal structures of the human 4-1BB receptor bound to its ligand 4-1BBL reveal covalent receptor dimerization as a potential signaling amplifier. J. Biol. Chem. 2018, 293, 9958–9969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Kim, A.M.J.; Murray, A.A.; Lim, S.-O. N-Glycosylation Facilitates 4-1BB Membrane Localization by Avoiding Its Multimerization. Cells 2022, 11, 162. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef] [PubMed]
- Van de Wall, S.; Santegoets, K.C.M.; van Houtum, E.J.H.; Büll, C.; Adema, G.J. Sialoglycans and Siglecs Can Shape the Tumor Immune Microenvironment. Trends Immunol. 2020, 41, 274–285. [Google Scholar] [CrossRef]
- Angata, T.; Tabuchi, Y.; Nakamura, K.; Nakamura, M. Siglec-15: An immune system Siglec conserved throughout vertebrate evolution. Glycobiology 2007, 17, 838–846. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef]
- Chen, X.; Dang, X.; Song, J.; Wang, G.; Liu, C.; Cui, L.; Huang, Z. N-glycosylation of Siglec-15 decreases its lysosome-dependent degradation and promotes its transportation to the cell membrane. Biochem. Biophys. Res. Commun. 2020, 533, 77–82. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.-A.; Zheleznyak, A.; Fang, Y.-F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a Marker of Self on Red Blood Cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Mawby, W.J.; Holmes, C.H.; Anstee, D.J.; Spring, F.A.; Tanner, M.J. Isolation and characterization of CD47 glycoprotein: A multi-spanning membrane protein which is the same as integrin-associated protein (IAP) and the ovarian tumour marker OA3. Biochem. J. 1994, 304 Pt 2, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T.; Discher, D.E. Post-translational regulation of expression and conformation of an immunoglobulin domain in yeast surface display. Biotechnol. Bioeng. 2006, 93, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Forgione, R.E.; Di Carluccio, C.; Guzmán-Caldentey, J.; Gaglione, R.; Battista, F.; Chiodo, F.; Manabe, Y.; Arciello, A.; Del Vecchio, P.; Fukase, K.; et al. Unveiling Molecular Recognition of Sialoglycans by Human Siglec-10. iScience 2020, 23, 101231. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.-G.; Steinle, A. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [Green Version]
- Andresen, L.; Skovbakke, S.L.; Persson, G.; Hagemann-Jensen, M.; Hansen, K.A.; Jensen, H.; Skov, S. 2-Deoxy d-Glucose Prevents Cell Surface Expression of NKG2D Ligands through Inhibition of N-Linked Glycosylation. J. Immunol. 2012, 188, 1847–1855. [Google Scholar] [CrossRef]
- Mellergaard, M.; Skovbakke, S.L.; Schneider, C.L.; Lauridsen, F.; Andresen, L.; Jensen, H.; Skov, S.; Mahesh, G.; Jeong, E.; Ng, F.S.; et al. N-Glycosylation of Asparagine 8 Regulates Surface Expression of Major Histocompatibility Complex Class I Chain-related Protein A (MICA) Alleles Dependent on Threonine 24. J. Biol. Chem. 2014, 289, 20078–20091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agresta, L.; Lehn, M.; Lampe, K.; Cantrell, R.; Hennies, C.; Szabo, S.; Wise-Draper, T.; Conforti, L.; Hoebe, K.; Janssen, E.M. CD244 represents a new therapeutic target in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e000245. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.H.; Boles, K.; Van Der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the Natural Killer and T Cell Immunoglobulin Superfamily Surface Protein, Is a Ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Margraf-Schönfeld, S.; Böhm, C.; Watzl, C. Glycosylation Affects Ligand Binding and Function of the Activating Natural Killer Cell Receptor 2B4 (CD244) Protein. J. Biol. Chem. 2011, 286, 24142–24149. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-N.; Lee, H.-H.; Hsu, J.L.; Yu, D.; Hung, M.-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 1–11. [Google Scholar] [CrossRef]
- Tan, W.; Jiang, P.; Zhang, W.; Hu, Z.; Lin, S.; Chen, L.; Li, Y.; Peng, C.; Li, Z.; Sun, A.; et al. Posttranscriptional regulation of de novo lipogenesis by glucose-induced O-GlcNAcylation. Mol. Cell 2021, 81, 1890–1904. [Google Scholar] [CrossRef]
- Nie, H.; Ju, H.; Fan, J.; Shi, X.; Cheng, Y.; Cang, X.; Zheng, Z.; Duan, X.; Yi, W. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat. Commun. 2020, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Chevet, E.; De Matteis, M.A.; Eskelinen, E.; Farhan, H. RNA, a new member in the glycan-club that gets exposed at the cell surface. Traffic 2021, 22, 362–363. [Google Scholar] [CrossRef]
- Disney, M.D. A glimpse at the glycoRNA world. Cell 2021, 184, 3080–3081. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Wang, R.; Jiao, S.; Wang, S.; Zhang, J.; Zhang, M. Identification of a monoclonal antibody that targets PD-1 in a manner requiring PD-1 Asn58 glycosylation. Commun. Biol. 2019, 2, 392. [Google Scholar] [CrossRef]
- Liu, J.; Wang, G.; Liu, L.; Wu, R.; Wu, Y.; Fang, C.; Zhou, X.; Jiao, J.; Gu, Y.; Zhou, H.; et al. Study of the interactions of a novel monoclonal antibody, mAb059c, with the hPD-1 receptor. Sci. Rep. 2019, 9, 17830. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Xu, Z.; Zhang, D.; Jiang, M.; Liu, K.; He, J.; Ma, D.; Ma, X.; Tan, S.; Gao, G.F.; et al. PD-1 N58-Glycosylation-Dependent Binding of Monoclonal Antibody Cemiplimab for Immune Checkpoint Therapy. Front. Immunol. 2022, 13, 826045. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-F.; Li, Z.; Ma, D.; Yu, Q. Small-molecule PD-L1 inhibitor BMS1166 abrogates the function of PD-L1 by blocking its ER export. OncoImmunology 2020, 9, 1831153. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sambrooks, C.; Shrimal, S.; Khodier, C.; Flaherty, D.P.; Rinis, N.; Charest, J.C.; Gao, N.; Zhao, P.; Wells, L.; Lewis, T.A.; et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol. 2016, 12, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Rinis, N.; Golden, J.E.; Marceau, C.D.; Carette, J.; Van Zandt, M.C.; Gilmore, R.; Contessa, J.N. Editing N-Glycan Site Occupancy with Small-Molecule Oligosaccharyltransferase Inhibitors. Cell Chem. Biol. 2018, 25, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Xia, L.; Hao, X.; Gan, F.; Bai, Y.; Zhang, C.; Mao, Y.; Zhu, Y.; Pu, Q.; Park, D.W.; et al. Targeting STT3A produces an anti-tumor effect in lung adenocarcinoma by blocking the MAPK and PI3K/AKT signaling pathway. Transl. Lung Cancer Res. 2022, 11, 1089–1107. [Google Scholar] [CrossRef]
- Shao, B.; Li, C.W.; Lim, S.O.; Sun, L.; Lai, Y.J.; Hou, J.; Liu, C.; Chang, C.W.; Qiu, Y.; Hsu, J.M.; et al. Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. Am. J. Cancer Res. 2018, 8, 1837–1846. [Google Scholar]
- Kim, B.; Sun, R.; Oh, W.; Kim, A.M.J.; Schwarz, J.R.; Lim, S.O. Saccharide analog, 2-deoxy-d-glucose enhances 4-1BB-mediated antitumor immunity via PD-L1 deglycosylation. Mol. Carcinog. 2020, 59, 691–700. [Google Scholar] [CrossRef]
- Iida, S.; Misaka, H.; Inoue, M.; Shibata, M.; Nakano, R.; Yamane-Ohnuki, N.; Wakitani, M.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated Therapeutic IgG1 Antibody Can Evade the Inhibitory Effect of Serum Immunoglobulin G on Antibody-Dependent Cellular Cytotoxicity through its High Binding to FcγRIIIa. Clin. Cancer Res. 2006, 12, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chong, G.; Zong, G.; Knorr, D.A.; Bournazos, S.; Aytenfisu, A.H.; Henry, G.K.; Ravetch, J.V.; MacKerell, A.D.; Wang, L.-X. Site-Selective Chemoenzymatic Modification on the Core Fucose of an Antibody Enhances Its Fcγ Receptor Affinity and ADCC Activity. J. Am. Chem. Soc. 2021, 143, 7828–7838. [Google Scholar] [CrossRef]
- Smith, A.; Zeng, W.; Lucas, S.; Grogan, B.; Blackmarr, A.; Wo, S.; Peterson, S.; Gardai, S. Abstract 1583: SEA-TGT is an empowered anti-TIGIT antibody that displays superior combinatorial activity with several therapeutic agents. Cancer Res. 2021, 81, 1583. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, L.; Yang, Q.; Li, F.; Zhu, M.; Yang, H.; Tan, T.; Wu, B.; Liu, M.; Xu, C.; Yin, J.; et al. The Glycosylation of Immune Checkpoints and Their Applications in Oncology. Pharmaceuticals 2022, 15, 1451. https://doi.org/10.3390/ph15121451
Zheng L, Yang Q, Li F, Zhu M, Yang H, Tan T, Wu B, Liu M, Xu C, Yin J, et al. The Glycosylation of Immune Checkpoints and Their Applications in Oncology. Pharmaceuticals. 2022; 15(12):1451. https://doi.org/10.3390/ph15121451
Chicago/Turabian StyleZheng, Linlin, Qi Yang, Feifei Li, Min Zhu, Haochi Yang, Tian Tan, Binghuo Wu, Mingxin Liu, Chuan Xu, Jun Yin, and et al. 2022. "The Glycosylation of Immune Checkpoints and Their Applications in Oncology" Pharmaceuticals 15, no. 12: 1451. https://doi.org/10.3390/ph15121451