
Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs

 , , , , and
, , , , and
Abstract
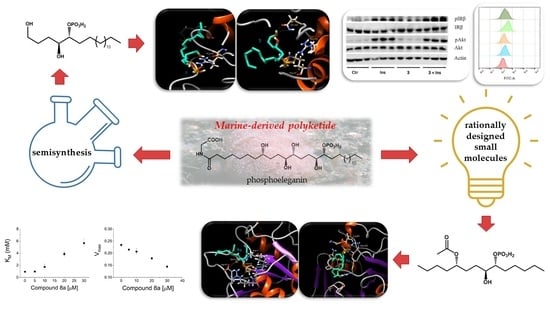
:
1. Introduction
2. Results
2.1. Oxidative Cleavage of Phosphoeleganin Affords Compounds 2 and 3
2.2. Synthetic Approach for Obtaining Compounds 4a–9b
2.3. Preliminary Screening on AR and PTP1B Enzymes of Compounds 2–4b and 7a–9b
2.4. Definition of 3 and 8a Mechanism of Action on PTP1B
2.5. Evaluation of 3 and 8a Interaction with PTP1B by In Silico Docking Analyses
2.6. Analysis of Insulin Signalling Pathway in C2C12 Cells Treated with Compound 3
2.7. Glucose Uptake Assay of Compound 3
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Compounds 2 and 3
4.3. Synthesis of the Series of Phosphoeleganin-Based Simplified Analogues 4a–9b
4.4. Enzymatic Assay
4.4.1. Enzymatic Assays with PTP1B
4.4.2. AR Enzymatic Assay
4.5. Determination of the IC50 Values and Reversibility Assay with PTP1B
4.6. Determination of the Mechanism of Inhibition on PTP1B
4.7. Cell Cultures
4.8. Insulin Signalling Pathway Analysis
4.9. Glucose Uptake Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chung-Davidson, Y.W.; Bussy, U.; Li, W. Recent Advances and Applications of Experimental Technologies in Marine Natural Product Research. Mar. Drugs 2015, 13, 2694–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casertano, M.; Menna, M.; Imperatore, C. The ascidian-derived metabolites with antimicrobial properties. Antibiotics 2020, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, D.; Luesch, H. Advances in exploring the therapeutic potential of marine natural products. Pharmacol. Res. 2019, 147, 104373. [Google Scholar] [CrossRef]
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. A Fragment Library of Natural Products and its Comparative Chemoinformatic Characterization. Mol. Inform. 2020, 39, e2000050. [Google Scholar] [CrossRef]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: What does it hold? Exp. Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Crane, E.A.; Gademann, K. Capturing Biological Activity in Natural Product Fragments by Chemical Synthesis. Angew. Chem. Int. Ed. 2016, 55, 3882–3902. [Google Scholar] [CrossRef] [Green Version]
- Maier, M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef] [Green Version]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.W.; Menna, M. Structure and Configuration of Phosphoeleganin, a Protein Tyrosine Phosphatase 1B Inhibitor from the Mediterranean Ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef]
- Luciano, P.; Imperatore, C.; Senese, M.; Aiello, A.; Casertano, M.; Guo, Y.W.; Menna, M. Assignment of the Absolute Configuration of Phosphoeleganin via Synthesis of Model Compounds. J. Nat. Prod. 2017, 80, 2118–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.; Imperatore, C.; Casertano, M.; Aiello, A.; Balestri, F.; Piazza, L.; Menna, M.; Del Corso, A.; Paoli, P. Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Mar. Drugs 2021, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Uetani, N.; Miranda-Saavedra, D.; Tremblay, M.L. PTP1B: A simple enzyme for a complex world. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 430–445. [Google Scholar] [CrossRef]
- Del Corso, A.; Cappiello, M.; Mura, U. From A Dull Enzyme to Something Else: Facts and Perspectives Regarding Aldose Reductase. Curr. Med. Chem. 2008, 15, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Dueymes, C.; Pirat, C.; Pascal, R. Facile synthesis of simple mono-alkyl phosphates from phosphoric acid and alcohols. Tetrahedron Lett. 2008, 49, 5300–5301. [Google Scholar] [CrossRef]
- Lira, L.M.; Vasilev, D.; Pilli, R.A.; Wessjohann, L.A. One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett. 2013, 54, 1690–1692. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Jia, Z.; Ye, Q.; Dinaut, A.N.; Wang, Q.; Waddleton, D.; Payette, P.; Ramachandran, C.; Kennedy, B.; Hum, G.; Taylor, S.D. Structure of protein tyrosine phosphatase 1B in complex with inhibitors bearing two phosphotyrosine mimetics. J. Med. Chem. 2001, 44, 4584–4594. [Google Scholar] [CrossRef]
- Sarmiento, M.; Zhao, Y.; Gordon, S.J.; Zhang, Z.Y. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1998, 273, 26368–26374. [Google Scholar] [CrossRef] [Green Version]
- Asante-Appiah, E.; Patel, S.; Dufresne, C.; Roy, P.; Wang, Q.; Patel, V.; Friesen, R.W.; Ramachandran, C.; Becker, J.W.; Leblanc, Y.; et al. The structure of PTP-1B in complex with a peptide inhibitor reveals an alternative binding mode for bisphosphonates. Biochemistry 2002, 41, 9043–9051. [Google Scholar] [CrossRef]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 23, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Pannifer, A.D.; Flint, A.J.; Tonks, N.K.; Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 1998, 273, 10454–10462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleasdale, J.E.; Ogg, D.; Palazuk, B.J.; Jacob, C.S.; Swanson, M.L.; Wang, X.Y.; Thompson, D.P.; Conradi, R.A.; Mathews, W.R.; Laborde, A.L.; et al. Small Molecule Peptidomimetics Containing a Novel Phosphotyrosine Bioisostere Inhibit Protein Tyrosine Phosphatase 1B and Augment Insulin Action. Biochemistry 2001, 40, 5642–5654. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Lee, S.Y.; Andersen, J.N.; Waters, S.; Shen, K.; Guo, X.L.; Moller, N.P.; Olefsky, J.M.; Lawrence, D.S.; Zhang, Z.Y. Cellular effects of small molecule PTP1B inhibitors on insulin signaling. Cellular Effects of Small Molecule PTP1B Inhibitors on Insulin Signaling. Biochemistry 2003, 42, 12792–12804. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Valadan, M.; Tartaglione, L.; Persico, M.; Ramunno, A.; Menna, M.; Casertano, M.; Dell’Aversano, C.; Singh, M.; d’Aulisio Garigliota, M.L.; et al. Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives. Int. J. Mol. Sci. 2020, 21, 1246. [Google Scholar] [CrossRef] [Green Version]
- Ottanà, R.; Paoli, P.; Lori, G.; Adornato, I.; Previti, S.; Naß, A.; Wolber, G.; Maccari, R. Design and evaluation of non-carboxylate 5-arylidene-2-thioxo-4-imidazolidinones as novel non-competitive inhibitors of protein tyrosine phosphatase 1B. Bioorg. Chem. 2019, 92, 103211. [Google Scholar] [CrossRef]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to D-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef] [Green Version]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) |
|---|---|
| Phosphoeleganin | 0.7 ± 0.1 |
| 3 | 6.7 ± 3.3 |
| 8a | 16.0 ± 2.0 |
| 9a | 83.0 ± 15.0 |
| 8b | 37.1 ± 6.5 |
| 9b | 63.0 ± 14.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casertano, M.; Genovese, M.; Piazza, L.; Balestri, F.; Del Corso, A.; Vito, A.; Paoli, P.; Santi, A.; Imperatore, C.; Menna, M. Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs. Pharmaceuticals 2022, 15, 325. https://doi.org/10.3390/ph15030325
Casertano M, Genovese M, Piazza L, Balestri F, Del Corso A, Vito A, Paoli P, Santi A, Imperatore C, Menna M. Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs. Pharmaceuticals. 2022; 15(3):325. https://doi.org/10.3390/ph15030325
Chicago/Turabian StyleCasertano, Marcello, Massimo Genovese, Lucia Piazza, Francesco Balestri, Antonella Del Corso, Alessio Vito, Paolo Paoli, Alice Santi, Concetta Imperatore, and Marialuisa Menna. 2022. "Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs" Pharmaceuticals 15, no. 3: 325. https://doi.org/10.3390/ph15030325