
Development and Biological Evaluation of the First Highly Potent and Specific Benzamide-Based Radiotracer [18F]BA3 for Imaging of Histone Deacetylases 1 and 2 in Brain

 , , , , ,
, , , , ,  , , , , and
, , , , and
Abstract
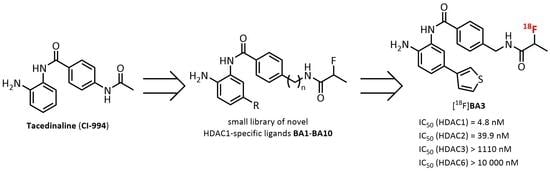
:
1. Introduction
2. Results and Discussion
2.1. Organic Chemistry
2.2. Determination of the In Vitro Inhibitory Potency
2.3. Precursor- and Radiosynthesis
2.3.1. Manual Radiosynthesis
2.3.2. Automated Radiosynthesis
2.4. Lipophilicity and Radiochemical Stability of [18F]BA3
2.5. In Vivo PET/MRI Studies of [18F]BA3
2.6. In Vivo Metabolism of [18F]BA3
2.7. Expression of HDAC1 in F98 and U251-MG Cells
2.8. The New HDAC Inhibitor BA3 Has Potential to Reduce the Proliferation of Cancer Cells
3. Materials and Methods
3.1. General
3.2. General Procedures
3.2.1. Suzuki Coupling A1
3.2.2. Nitro Reduction with Hydrogen A2a
3.2.3. Nitro Reduction with Sodium Dithionite A2b
3.2.4. Amide Coupling A3
3.2.5. Boc Cleavage A4
3.3. Compounds
3.3.1. Synthesis of tert-butyl (4-bromo-2-nitrophenyl)carbamate (1)
3.3.2. Synthesis of tert-butyl [2-nitro-4-(thiophen-2-yl)phenyl]carbamate (2a)
3.3.3. Synthesis of tert-butyl [2-nitro-4-(thiophen-3-yl)phenyl]carbamate (2b)
3.3.4. Synthesis of tert-butyl (4′-fluoro-3-nitro-[1,1′-biphenyl]-4-yl)carbamate (2c)
3.3.5. Synthesis of tert-butyl [4-(furan-2-yl)-2-nitrophenyl]carbamate (2d)
3.3.6. Synthesis of tert-butyl [4-(furan-3-yl)-2-nitrophenyl]carbamate (2e)
3.3.7. Synthesis of tert-butyl [2-amino-4-(thiophen-2-yl)phenyl]carbamate (3a)
3.3.8. Synthesis of tert-butyl [2-amino-4-(thiophen-3-yl)phenyl]carbamate (3b)
3.3.9. Synthesis of tert-butyl (3-amino-4′-fluoro-[1,1′-biphenyl]-4-yl)carbamate (3c)
3.3.10. Synthesis of tert-butyl [2-amino-4-(furan-2-yl)phenyl]carbamate (3d)
3.3.11. Synthesis of tert-butyl [2-amino-4-(furan-3-yl)phenyl]carbamate (3e)
3.3.12. Synthesis of tert-butyl (2-{4-[(2-fluoropropanamido)methyl]benzamido}-4-[thiophen-2-yl]phenyl)carbamate (4a)
3.3.13. Synthesis of tert-butyl (2-{4-[(2-fluoropropanamido)methyl]benzamido}-4-[thiophen-3-yl]phenyl)carbamate (4b)
3.3.14. Synthesis of tert-butyl (4′-fluoro-3-{4-[(2-fluoropropanamido)methyl]benzamido}-[1,1′-biphenyl]-4-yl)carbamate (4c)
3.3.15. Synthesis of tert-butyl (2-{4-[(2-fluoropropanamido)methyl]benzamido}-4-[furan-2-yl]phenyl)carbamate (4d)
3.3.16. Synthesis of tert-butyl (2-{4-[(2-fluoropropanamido)methyl]benzamido}-4-[furan-3-yl]phenyl)carbamate (4e)
3.3.17. Synthesis of tert-butyl {2-[4-(2-fluoropropanamido)benzamido]-4-(thiophen-2-yl)phenyl}carbamate (5a)
3.3.18. Synthesis of tert-butyl {2-[4-(2-fluoropropanamido)benzamido]-4-(thiophen-3-yl)phenyl}carbamate (5b)
3.3.19. Synthesis of tert-butyl {4′-fluoro-3-[4-(2-fluoropropanamido)benzamido]-[1,1′-biphenyl]-4-yl}carbamate (5c)
3.3.20. Synthesis of tert-butyl {2-[4-(2-fluoropropanamido)benzamido]-4-(furan-2-yl)phenyl}carbamate (5d)
3.3.21. Synthesis of tert-butyl {2-[4-(2-fluoropropanamido)benzamido]-4-(furan-3-yl)phenyl}carbamate (5e)
3.3.22. Synthesis of 2-fluoropropanoyl chloride (6)
3.3.23. Synthesis of 4-[(2-fluoropropanamido)methyl]benzoic acid (7)
3.3.24. Synthesis of 4-(2-fluoropropanamido)benzoic acid (8)
3.3.25. Synthesis of tert-butyl (2-{4-[(2-bromopropanamido)methyl]benzamido}-4-[thiophen-3-yl]phenyl)carbamate (9)
3.3.26. Synthesis of N-[2-amino-5-(thiophen-2-yl)phenyl]-4-[(2-fluoropropanamido)methyl]benzamide (BA1)
3.3.27. Synthesis of N-[2-amino-5-(thiophen-2-yl)phenyl]-4-(2-fluoropropanamido)benzamide (BA2)
3.3.28. Synthesis of N-[2-amino-5-(thiophen-3-yl)phenyl]-4-[(2-fluoropropanamido)methyl]benzamide (BA3)
3.3.29. Synthesis of N-[2-amino-5-(thiophen-3-yl)phenyl]-4-(2-fluoropropanamido)benzamide (BA4)
3.3.30. Synthesis of N-(4-amino-4′-fluoro-[1,1′-biphenyl]-3-yl)-4-[(2-fluoropropanamido)methyl]benzamide (BA5)
3.3.31. Synthesis of N-(4-amino-4′-fluoro-[1,1′-biphenyl]-3-yl)-4-(2-fluoropropanamido)benzamide (BA6)
3.3.32. Synthesis of N-[2-amino-5-(furan-2-yl)phenyl]-4-[(2-fluoropropanamido)methyl]benzamide (BA7)
3.3.33. Synthesis of N-[2-amino-5-(furan-2-yl)phenyl]-4-(2-fluoropropanamido)benzamide (BA8)
3.3.34. Synthesis of N-[2-amino-5-(furan-3-yl)phenyl]-4-[(2-fluoropropanamido)methyl]benzamide (BA9)
3.3.35. Synthesis of N-[2-amino-5-(furan-3-yl)phenyl]-4-(2-fluoropropanamido)benzamide (BA10)
3.4. Radiosynthesis
3.4.1. Manual Radiosynthesis of [18F]BA3
3.4.2. Automated Radiosynthesis of [18F]BA3
3.4.3. Quality Control and Analyses
3.4.4. Determination of Radiochemical Stability and Lipophilicity (logD7.4)
3.4.5. Inhibition Assay for HDAC1-3 and HDAC6
3.5. Biological Experiments
3.5.1. Metabolite Analysis
3.5.2. Cell Culture
3.5.3. Immunofluorescence Staining
3.5.4. MTS Assay
3.5.5. PET Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tago, T.; Toyohara, J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules 2018, 23, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [Green Version]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Wang, D.F.; Helquist, P.; Wiech, N.L.; Wiest, O. Toward selective histone deacetylase inhibitor design: Homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J. Med. Chem. 2005, 48, 6936–6947. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat. Commun. 2020, 11, 3252. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Wang, X.Q.; Bai, H.M.; Li, S.T.; Sun, H.; Min, L.Z.; Tao, B.B.; Zhong, J.; Li, B. Knockdown of HDAC1 expression suppresses invasion and induces apoptosis in glioma cells. Oncotarget 2017, 8, 48027–48040. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Ryu, H.W.; Won, H.R.; Kwon, S.H. Advances in epigenetic glioblastoma therapy. Oncotarget 2017, 8, 18577–18589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Jenke, R.; Ressing, N.; Hansen, F.K.; Aigner, A.; Buch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol. Ther. 2021, 220, 107721. [Google Scholar] [CrossRef]
- Camphausen, K.; Burgan, W.; Cerra, M.; Oswald, K.A.; Trepel, J.B.; Lee, M.J.; Tofilon, P.J. Enhanced radiation-induced cell killing and prolongation of gammaH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res. 2004, 64, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Camphausen, K.; Scott, T.; Sproull, M.; Tofilon, P.J. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin. Cancer Res. 2004, 10, 6066–6071. [Google Scholar] [CrossRef] [Green Version]
- Diss, E.; Nalabothula, N.; Nguyen, D.M.; Chang, E.T.; Kwok, Y.; Carrier, F. VorinostatSAHA Promotes Hyper-Radiosensitivity in Wild Type p53 Human Glioblastoma Cells. JSM Clin. Oncol. Res. 2014, 2, 1004. [Google Scholar]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, U.; Tong, W.P.; Gelovani, J.G.; Alauddin, M.M. Radiosynthesis of 6-([18F]fluoroacetamido)-1-hexanoicanilide ([18F]FAHA) for PET imaging of histone deacetylase (HDAC). J. Label. Compd. Radiopharm. 2006, 49, 997–1006. [Google Scholar] [CrossRef]
- Reid, A.E.; Hooker, J.; Shumay, E.; Logan, J.; Shea, C.; Kim, S.W.; Collins, S.; Xu, Y.; Volkow, N.; Fowler, J.S. Evaluation of 6-([18F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. Nucl. Med. Biol. 2009, 36, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.H.; Tian, M.; Hinz, R.; Young, D.; Shavrin, A.; Mukhapadhyay, U.; Flores, L.G.; Balatoni, J.; Soghomonyan, S.; Jeong, H.J.; et al. Imaging epigenetic regulation by histone deacetylases in the brain using PET/MRI with 18F-FAHA. Neuroimage 2013, 64, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Cho, S.J.; Yu, L.; Hudson, R.H.; Luyt, L.; Pin, C.; Kovacs, M.; Koropatnick, J.; Lee, T.-Y. Evaluation of 6-([18F] fluoroacetamido)-1-hexanoic-anilide (18F-FAHA) as imaging probe in tumor xenograft mice model. SPIE Med. Imaging 2016, 9788, 978814. [Google Scholar] [CrossRef]
- Fukumitsu, N.; Yeh, S.H.; Flores Ii, L.G.; Mukhopadhyay, U.; Young, D.; Ogawa, K.; Jeong, H.J.; Tong, W.; Gelovani, J.G. In Vivo 6-([18F]Fluoroacetamido)-1-hexanoicanilide PET Imaging of Altered Histone Deacetylase Activity in Chemotherapy-Induced Neurotoxicity. Contrast Media Mol. Imaging 2018, 2018, 3612027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, S.H.; Lin, M.H.; Leo Garcia, F., II; Mukhopadhyay, U.; Young, D.; Ogawa, K.; Jeong, J.H.; Tong, W.; Gelovani, J.G.; Fukumitsu, N. In Vivo Evaluation of the Combined Anticancer Effects of Cisplatin and SAHA in Nonsmall Cell Lung Carcinoma Using [18F]FAHA and [18F]FDG PET/CT Imaging. Mol. Imaging 2021, 2021, 6660358. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Wang, C.; Van de Bittner, G.C.; Neelamegam, R.; Takakura, W.R.; Karunakaran, A.; Wey, H.Y.; Reis, S.A.; Gale, J.; Zhang, Y.L.; et al. PET imaging demonstrates histone deacetylase target engagement and clarifies brain penetrance of known and novel small molecule inhibitors in rat. ACS Chem. Neurosci. 2014, 5, 1055–1062. [Google Scholar] [CrossRef]
- Wey, H.Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [Green Version]
- Skipper, P.L.; Tannenbaum, S.R.; Thilly, W.G.; Furth, E.E.; Bishop, W.W. Mutagenicity of Hydroxamic Acids and the Probable Involvement of Carbamoylation. Cancer Res. 1980, 40, 4704–4708. [Google Scholar]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Seo, Y.J.; Kang, Y.; Muench, L.; Reid, A.; Caesar, S.; Jean, L.; Wagner, F.; Holson, E.; Haggarty, S.J.; Weiss, P.; et al. Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem. Neurosci. 2014, 5, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [11C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.C.; Liu, J.; et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone deacetylase inhibitors: From bench to clinic. J. Med. Chem. 2008, 51, 1505–1529. [Google Scholar] [CrossRef]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef]
- Wambua, M.K.; Nalawansha, D.A.; Negmeldin, A.T.; Pflum, M.K. Mutagenesis studies of the 14 A internal cavity of histone deacetylase 1: Insights toward the acetate-escape hypothesis and selective inhibitor design. J. Med. Chem. 2014, 57, 642–650. [Google Scholar] [CrossRef]
- Rogers, G.A.; Stone-Elander, S.; Ingvar, M.; Eriksson, L.; Parsons, S.M.; Widén, L. 18F-labelled vesamicol derivatives: Syntheses and preliminary in vivo small animal positron emission tomography evaluation. Nucl. Med. Biol. 1994, 21, 219–230. [Google Scholar] [CrossRef]
- Sorger, D.; Scheunemann, M.; Grossmann, U.; Fischer, S.; Vercouille, J.; Hiller, A.; Wenzel, B.; Roghani, A.; Schliebs, R.; Brust, P.; et al. A new 18F-labeled fluoroacetylmorpholino derivative of vesamicol for neuroimaging of the vesicular acetylcholine transporter. Nucl. Med. Biol. 2008, 35, 185–195. [Google Scholar] [CrossRef]
- Hu, K.Z.; Wang, H.; Huang, T.; Tang, G.; Liang, X.; He, S.; Tang, X. Synthesis and biological evaluation of N-(2-[(18)F]Fluoropropionyl)-L-methionine for tumor imaging. Nucl. Med. Biol. 2013, 40, 926–932. [Google Scholar] [CrossRef]
- Hu, K.; Tang, X.; Tang, G.; Yao, S.; Yao, B.; Wang, H.; Nie, D.; Liang, X.; Tang, C.; He, S. 18F-FP-PEG2-beta-Glu-RGD2: A Symmetric Integrin alphavbeta3-Targeting Radiotracer for Tumor PET Imaging. PLoS ONE 2015, 10, e0138675. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Tang, G.; Gao, S.; Liu, S.; Wen, F.; Yao, B.; Nie, D. Radiosynthesis and preliminary biological evaluation of N-(2-[18F]fluoropropionyl)-L-glutamine as a PET tracer for tumor imaging. Oncotarget 2016, 7, 34100–34111. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Q.; Dong, W.; Yang, L.; Lu, K.; Guo, X.; Liu, H.; Wei, H.; Cheng, Y.; Wu, Z.; et al. Radiosynthesis and evaluation of N(5)-(2-(18)F-fluoropropanyl) ornithine as a potential agent for tumor PET imaging. Nucl. Med. Biol. 2021, 94–95, 98–105. [Google Scholar] [CrossRef]
- Demont, E.H.; Bamborough, P.; Chung, C.W.; Craggs, P.D.; Fallon, D.; Gordon, L.J.; Grandi, P.; Hobbs, C.I.; Hussain, J.; Jones, E.J.; et al. 1,3-Dimethyl Benzimidazolones Are Potent, Selective Inhibitors of the BRPF1 Bromodomain. ACS Med. Chem. Lett. 2014, 5, 1190–1195. [Google Scholar] [CrossRef]
- Methot, J.L.; Hoffman, D.M.; Witter, D.J.; Stanton, M.G.; Harrington, P.; Hamblett, C.; Siliphaivanh, P.; Wilson, K.; Hubbs, J.; Heidebrecht, R.; et al. Delayed and Prolonged Histone Hyperacetylation with a Selective HDAC1/HDAC2 Inhibitor. ACS Med. Chem. Lett. 2014, 5, 340–345. [Google Scholar] [CrossRef] [Green Version]
- Witter, D.J.; Harrington, P.; Wilson, K.J.; Chenard, M.; Fleming, J.C.; Haines, B.; Kral, A.M.; Secrist, J.P.; Miller, T.A. Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Jiang, Y.; Wu, J.; Inks, E.S.; Chou, C.J.; Gao, S.; Hou, J.; Ding, Q.; Li, J.; et al. Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: Design, structure-activity relationship and anti-tumor activity study. Eur. J. Chem. 2017, 134, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zang, J.; Gao, Q.; Liang, X.; Ding, Q.; Li, X.; Xu, W.; Chou, C.J.; Zhang, Y. Design, synthesis and anti-tumor activity study of novel histone deacetylase inhibitors containing isatin-based caps and o-phenylenediamine-based zinc binding groups. Bioorg. Med. Chem. 2017, 25, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- La, M.T.; Jeong, B.H.; Kim, H.K. Design and Synthesis of Novel N-(2-aminophenyl)benzamide Derivatives as Histone Deacetylase Inhibitors and Their Antitumor Activity Study. Bull. Korean Chem. Soc. 2021, 42, 740–743. [Google Scholar] [CrossRef]
- Kilchmann, F.; Marcaida, M.J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gonczy, P.; Reymond, J.L. Discovery of a Selective Aurora A Kinase Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 7188–7211. [Google Scholar] [CrossRef]
- Schäker-Hübner, L.; Haschemi, R.; Büch, T.; Kraft, F.B.; Brumme, B.; Schöler, A.; Jenke, R.; Meiler, J.; Aigner, A.; Bendas, G.; et al. Balancing Histone Deacetylase (HDAC) Inhibition and Drug-likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. ChemMedChem 2022, e202100755. [Google Scholar] [CrossRef] [PubMed]
- Bratteby, K.; Shalgunov, V.; Battisti, U.M.; Petersen, I.N.; van den Broek, S.L.; Ohlsson, T.; Gillings, N.; Erlandsson, M.; Herth, M.M. Insights into Elution of Anion Exchange Cartridges: Opening the Path toward Aliphatic (18)F-Radiolabeling of Base-Sensitive Tracers. ACS Pharmacol. Transl. Sci. 2021, 4, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.P.; Terpinski, J.; Lawrynowicz, W. Anhydrous Tetrabutylammonium Fluoride—A Mild but Highly Efficient Source of Nucleophilic Fluoride-Ion. J. Org. Chem. 1984, 49, 3216–3219. [Google Scholar] [CrossRef]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef]
- Waterhouse, R.N. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef]
- Lindemann, M.; Moldovan, R.P.; Hinz, S.; Deuther-Conrad, W.; Gundel, D.; Dukic-Stefanovic, S.; Toussaint, M.; Teodoro, R.; Juhl, C.; Steinbach, J.; et al. Development of a Radiofluorinated Adenosine A2B Receptor Antagonist as Potential Ligand for PET Imaging. Int. J. Mol. Sci. 2020, 21, 3197. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Hiranaka, S.; Tega, Y.; Higuchi, K.; Kurosawa, T.; Deguchi, Y.; Arata, M.; Ito, A.; Yoshida, M.; Nagaoka, Y.; Sumiyoshi, T. Design, Synthesis, and Blood-Brain Barrier Transport Study of Pyrilamine Derivatives as Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2018, 9, 884–888. [Google Scholar] [CrossRef]
- Yakubovich, A.Y.; Gitel, P.O. Some properties of α-fluoronitriles. J. Gen. Chem. USSR 1966, 36, 889–891. [Google Scholar]
- Banks, J.W.; O’Hagan, D. The enzymatic resolution of an α-fluoroamide by an acylase. J. Fluor. Chem. 2000, 102, 235–238. [Google Scholar] [CrossRef]
- Rotering, S.; Franke, K.; Zessin, J.; Brust, P.; Fuchtner, F.; Fischer, S.; Steinbach, J. Convenient recycling and reuse of bombarded [18O]H2O for the production and the application of [18F]F−. Appl. Radiat. Isot. 2015, 101, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, M.; Hinz, S.; Deuther-Conrad, W.; Namasivayam, V.; Dukic-Stefanovic, S.; Teodoro, R.; Toussaint, M.; Kranz, M.; Juhl, C.; Steinbach, J.; et al. Radiosynthesis and in vivo evaluation of a fluorine-18 labeled pyrazine based radioligand for PET imaging of the adenosine A2B receptor. Bioorg. Med. Chem. 2018, 26, 4650–4663. [Google Scholar] [CrossRef] [PubMed]
- Schäker-Hübner, L.; Warstat, R.; Ahlert, H.; Mishra, P.; Kraft, F.B.; Schliehe-Diecks, J.; Scholer, A.; Borkhardt, A.; Breit, B.; Bhatia, S.; et al. 4-Acyl Pyrrole Capped HDAC Inhibitors: A New Scaffold for Hybrid Inhibitors of BET Proteins and Histone Deacetylases as Antileukemia Drug Leads. J. Med. Chem. 2021, 64, 14620–14646. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Dequiedt, F.; Verdin, E.; Jung, M. Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal. Biochem. 2003, 319, 42–48. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Enzymes | Localization | Expression |
|---|---|---|---|
| I | HDAC1, 2, 3, 8 | Nucleus | Ubiquitous |
| IIa | HDAC4, 5, 7, 9 | Nucleus and cytoplasm | Tissue specific |
| IIb | HDAC6, 10 | Cytoplasm | Tissue specific |
| III | Sirtuins 1–7 | Variable | Variable |
| IV | HDAC11 | Nucleus and cytoplasm | Ubiquitous |
| Compound | R | n | IC50 [nM] | |||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | |||
| BA1 | 2-thienyl | 1 | 4.8 ± 0.4 | 64.3 ± 4.5 | 2300 ± 190 | >10,000 a |
| BA2 | 2-thienyl | 0 | 8.3 ± 1.4 | 33.5 ± 1.5 | >1110 b | >10,000 a |
| BA3 | 3-thienyl | 1 | 4.8 ± 0.6 | 39.9 ± 3.2 | >1110 b | >10,000 a |
| BA4 | 3-thienyl | 0 | 11.2 ± 2.4 | 38.1 ± 4.5 | >3330 b | >10,000 a |
| BA5 | 4-fluorophenyl | 1 | 10.2 ± 0.4 | 32.2 ± 5.5 | 1300 ± 70 | >10,000 a |
| BA6 | 4-fluorophenyl | 0 | 14.9 ± 0.4 | 56.6 ± 0.6 | >1110 b | >10,000 a |
| BA7 | 2-furanyl | 1 | 20.4 ± 0.3 | 69.1 ± 1.2 | 3800 ± 180 | >10,000 a |
| BA8 | 2-furanyl | 0 | 24.2 ± 2.9 | 82.1 ± 0.1 | 4200 ± 80 | >10,000 a |
| BA9 | 3-furanyl | 1 | 19.8 ± 0.4 | 56.3 ± 0.2 | 2400 ± 250 | >10,000 a |
| BA10 | 3-furanyl | 0 | 17.0 ± 1.2 | 47.7 ± 2.9 | 3300 ± 480 | >10,000 a |
| vorinostat | - | - | 124 ± 15 | 197 ± 14 | 129 ± 7 | 22 ± 3 |
| entinostat c | - | - | 426 ± 59 | 354 ± 43 | 311 ± 10 | >10,000 a |
| tacedinaline | - | - | 636 ± 114 | 696 ± 11 | 263 ± 31 | >10,000 a |
| Cell Viability [%] (Vehicle = 100%) | ||
|---|---|---|
| HDAC Inhibitor (conc.) | U251-MG | F98 |
| BA3 (< 50 µM a) | 63.6 ± 20.7 | 36.3 ± 7.6 |
| vorinostat (50 µM) | 36.9 ± 7.6 | 24.4 ± 7.9 |
| entinostat (50 µM) | 55.0 ± 11.2 | 27.5 ± 4.4 |
| tacedinaline (50 µM) | 28.5 ± 3.0 | 27.1 ± 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clauß, O.; Schäker-Hübner, L.; Wenzel, B.; Toussaint, M.; Deuther-Conrad, W.; Gündel, D.; Teodoro, R.; Dukić-Stefanović, S.; Ludwig, F.-A.; Kopka, K.; et al. Development and Biological Evaluation of the First Highly Potent and Specific Benzamide-Based Radiotracer [18F]BA3 for Imaging of Histone Deacetylases 1 and 2 in Brain. Pharmaceuticals 2022, 15, 324. https://doi.org/10.3390/ph15030324
Clauß O, Schäker-Hübner L, Wenzel B, Toussaint M, Deuther-Conrad W, Gündel D, Teodoro R, Dukić-Stefanović S, Ludwig F-A, Kopka K, et al. Development and Biological Evaluation of the First Highly Potent and Specific Benzamide-Based Radiotracer [18F]BA3 for Imaging of Histone Deacetylases 1 and 2 in Brain. Pharmaceuticals. 2022; 15(3):324. https://doi.org/10.3390/ph15030324
Chicago/Turabian StyleClauß, Oliver, Linda Schäker-Hübner, Barbara Wenzel, Magali Toussaint, Winnie Deuther-Conrad, Daniel Gündel, Rodrigo Teodoro, Sladjana Dukić-Stefanović, Friedrich-Alexander Ludwig, Klaus Kopka, and et al. 2022. "Development and Biological Evaluation of the First Highly Potent and Specific Benzamide-Based Radiotracer [18F]BA3 for Imaging of Histone Deacetylases 1 and 2 in Brain" Pharmaceuticals 15, no. 3: 324. https://doi.org/10.3390/ph15030324