
Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery

 ,
, 
 , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Development of Isatin Derivatives as Promising Therapeutic Agents
2.1. Anticancer Activity of Isatin
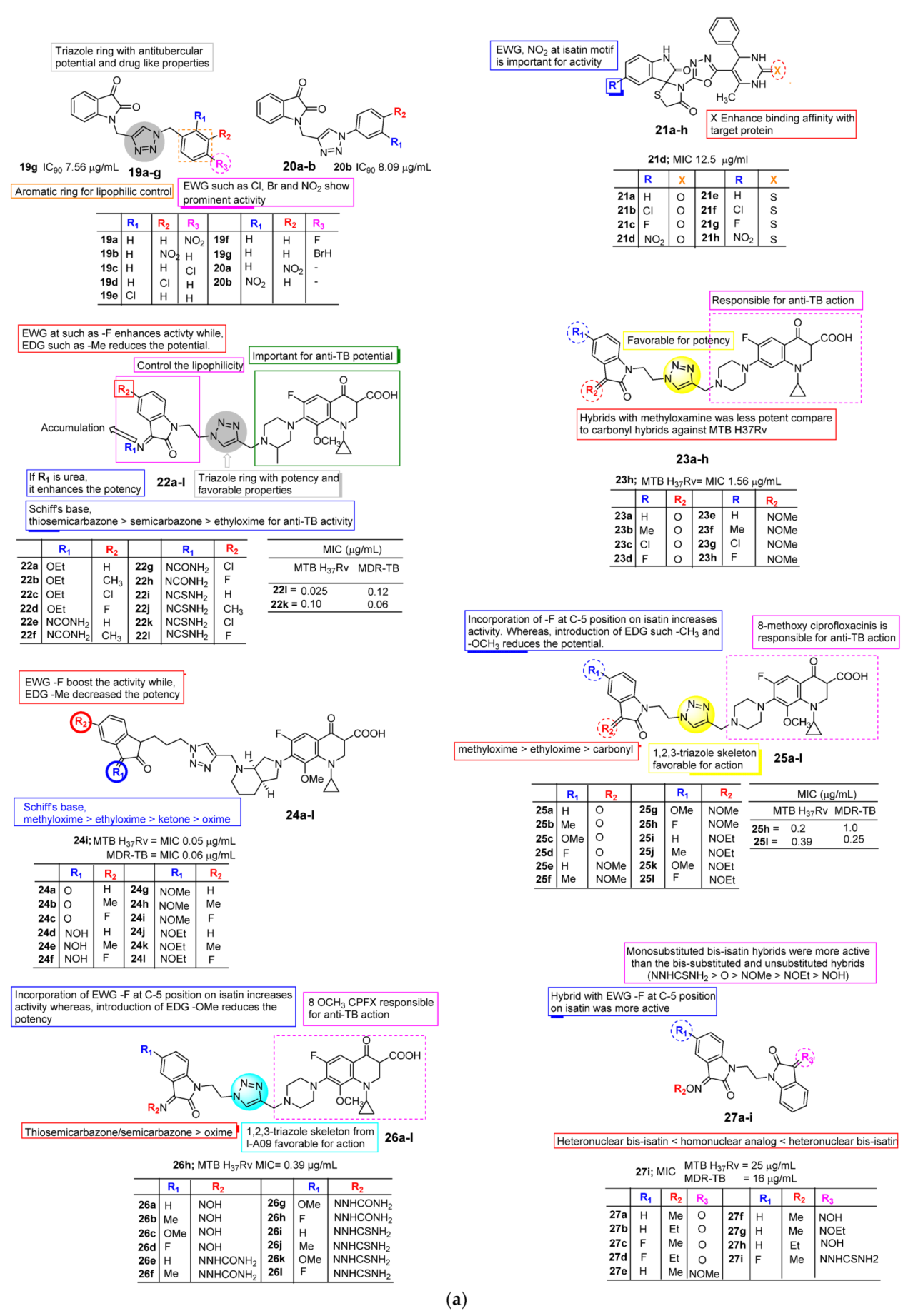
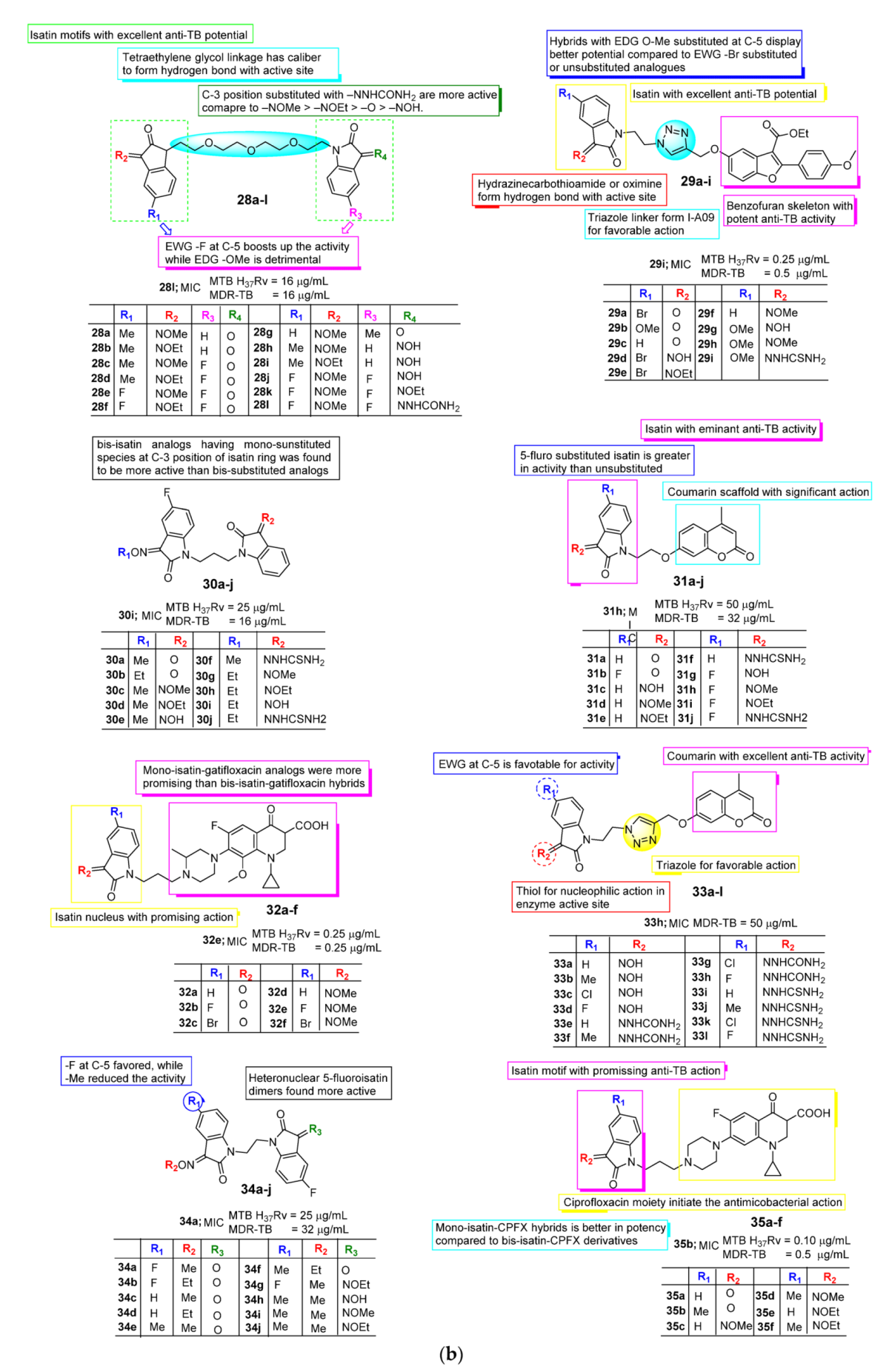
2.2. Anti-TB Activity of Isatin
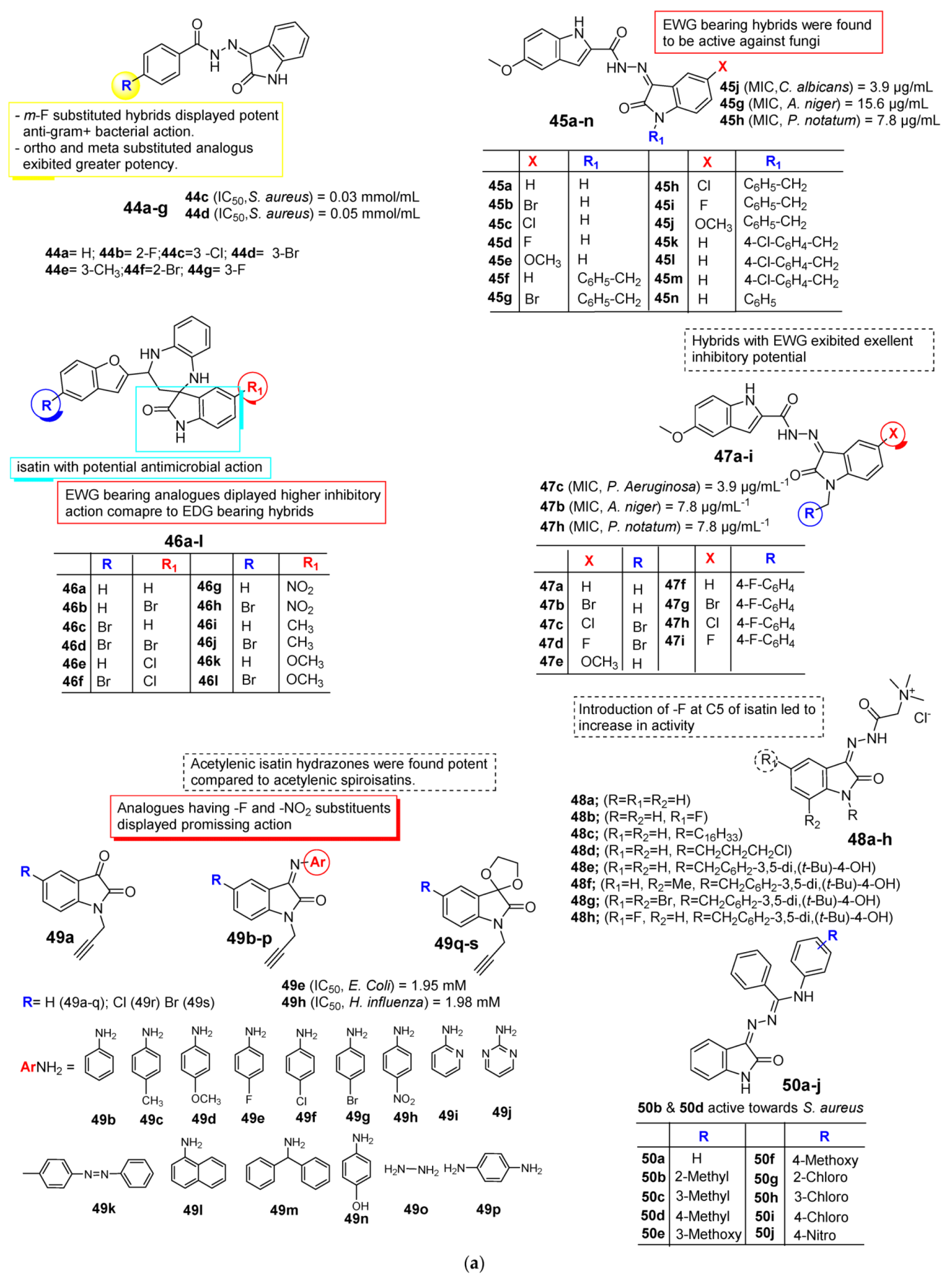
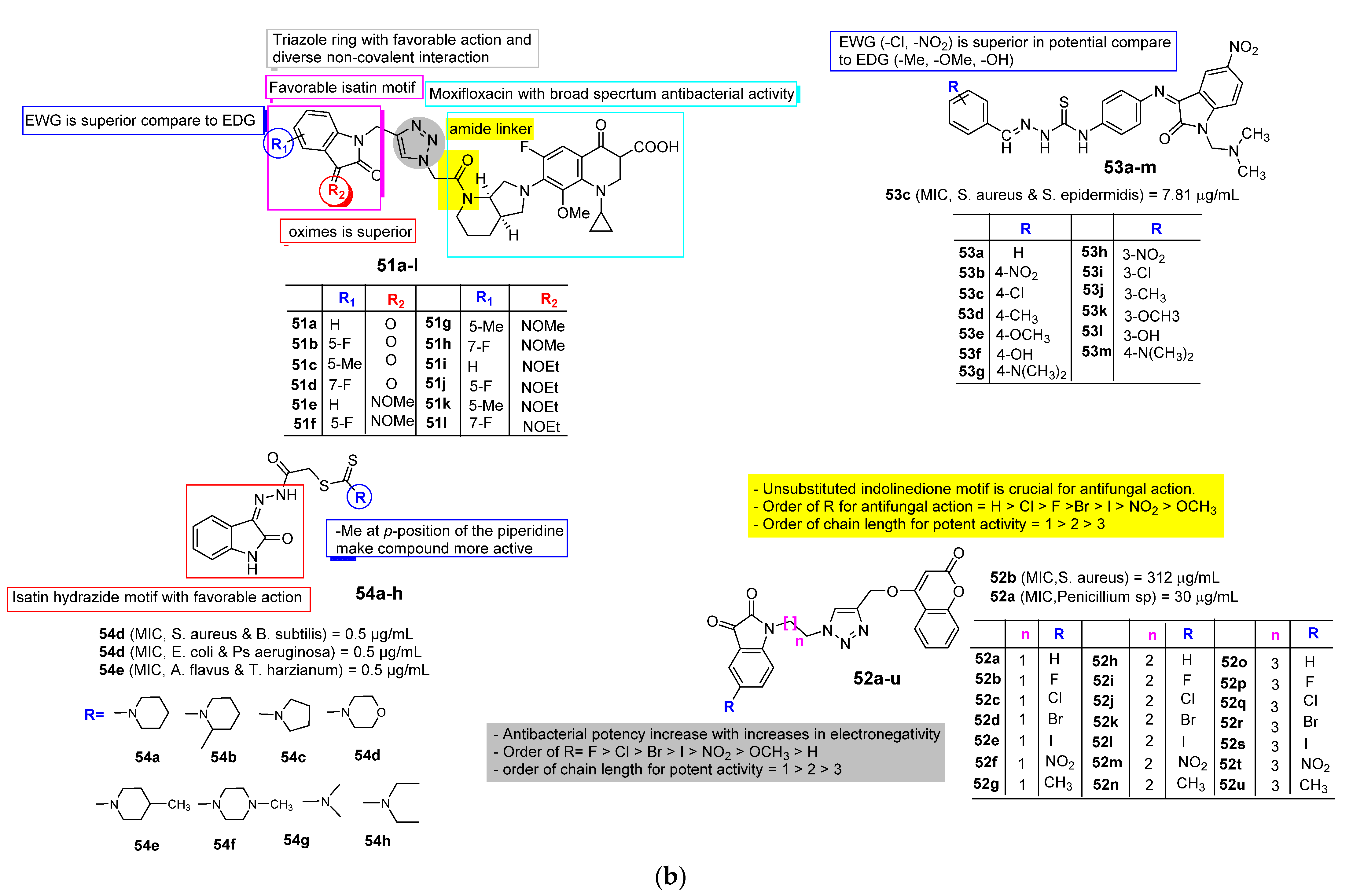
2.3. Isatin as Antimicrobial Agent
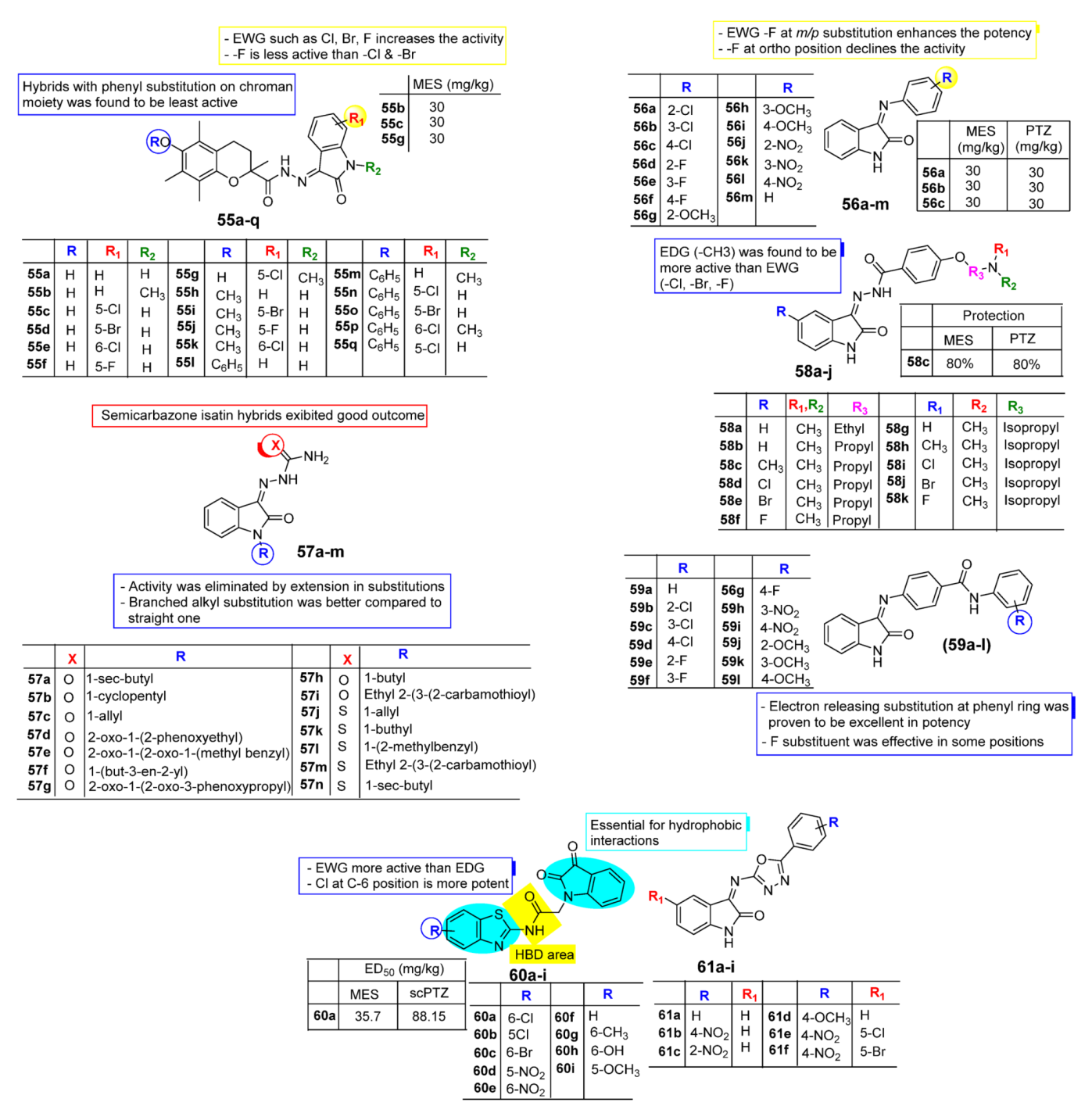
2.4. Isatin as Anti-Convulsant Agent
2.5. Antioxidant Potential of Isatin
2.6. Anti-Inflammatory Potential of Isatin
2.7. Antidiabetic Profile of Isatin
2.8. Isatin as Anti-HIV Agent
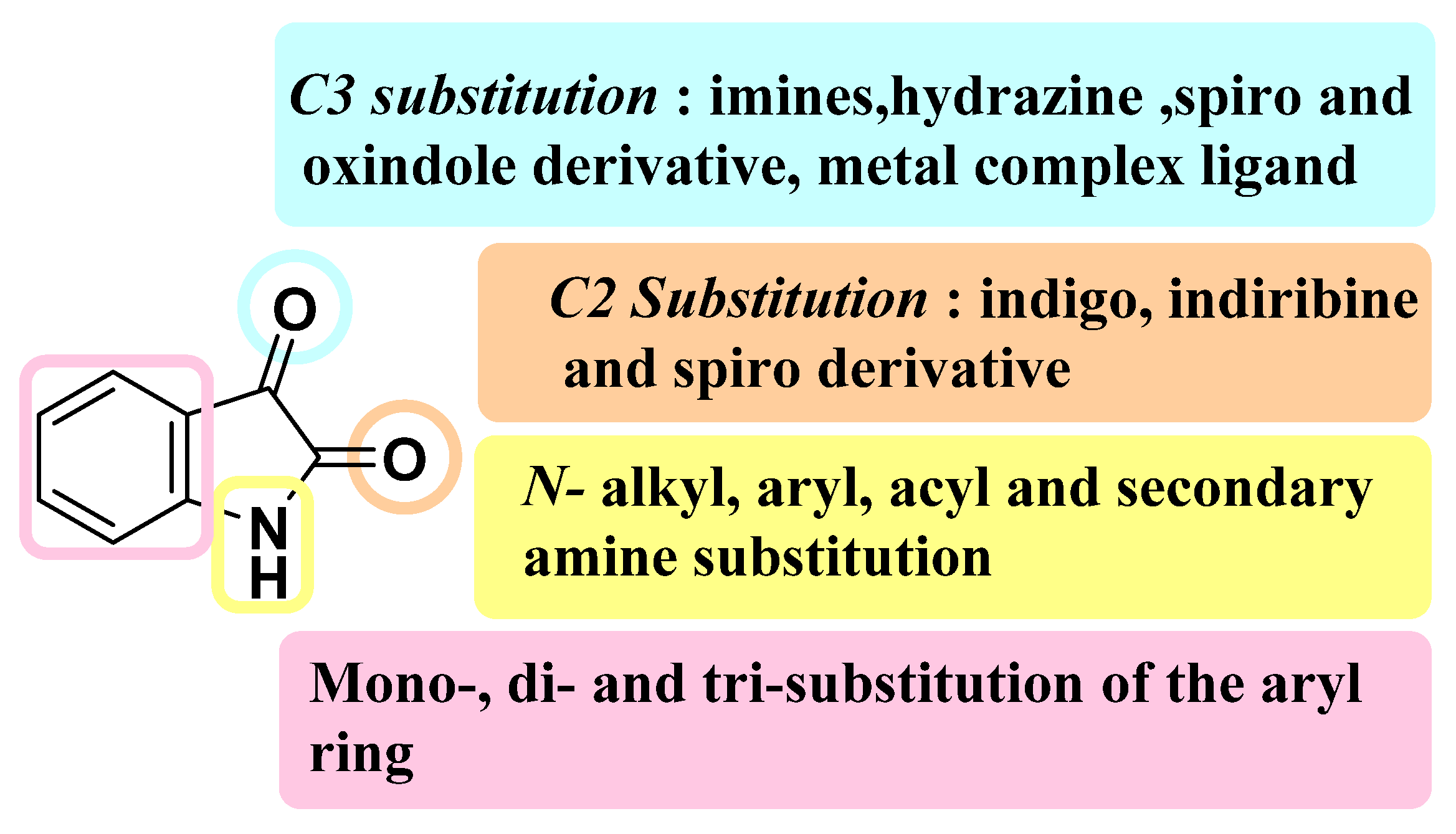
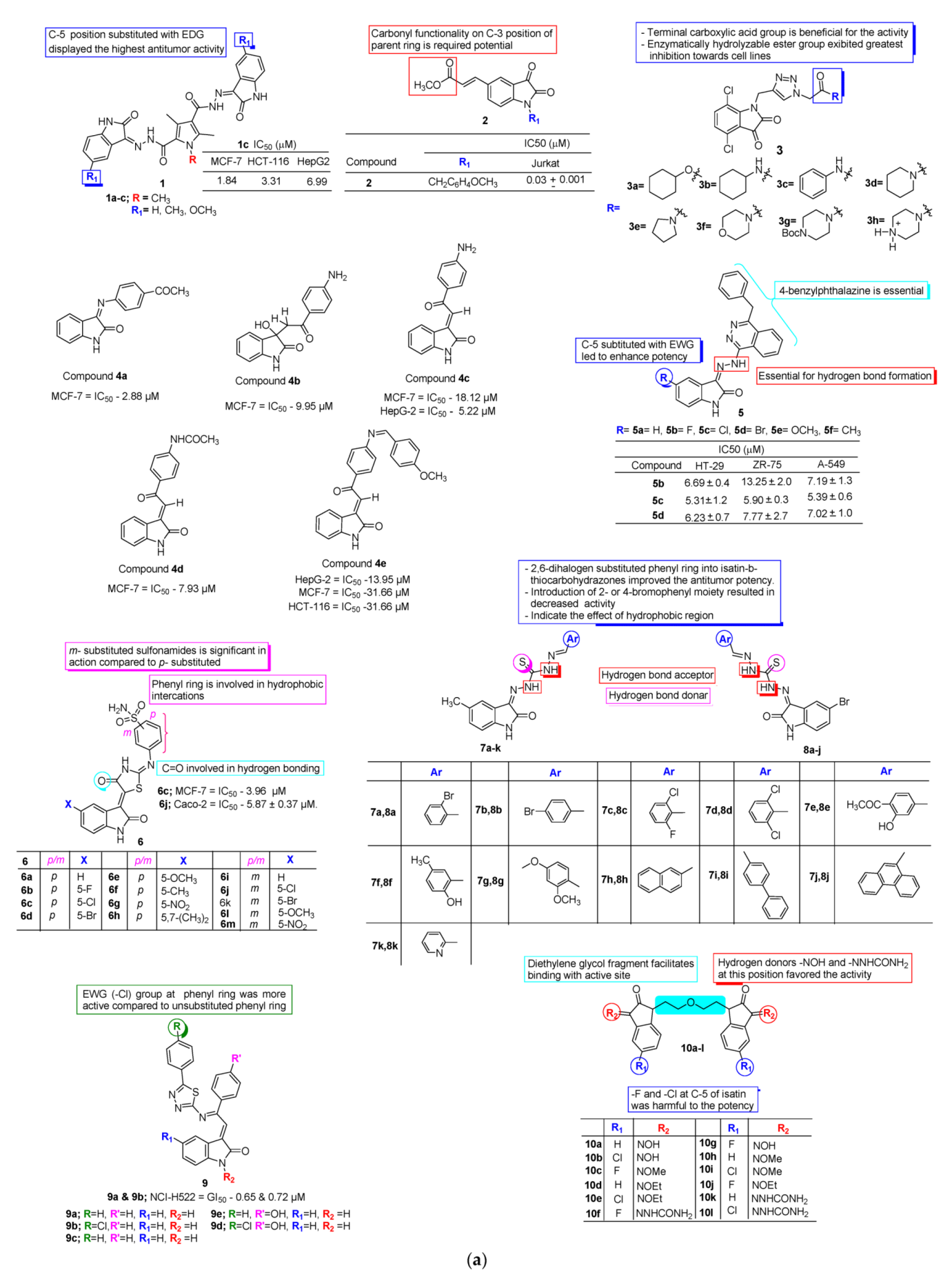
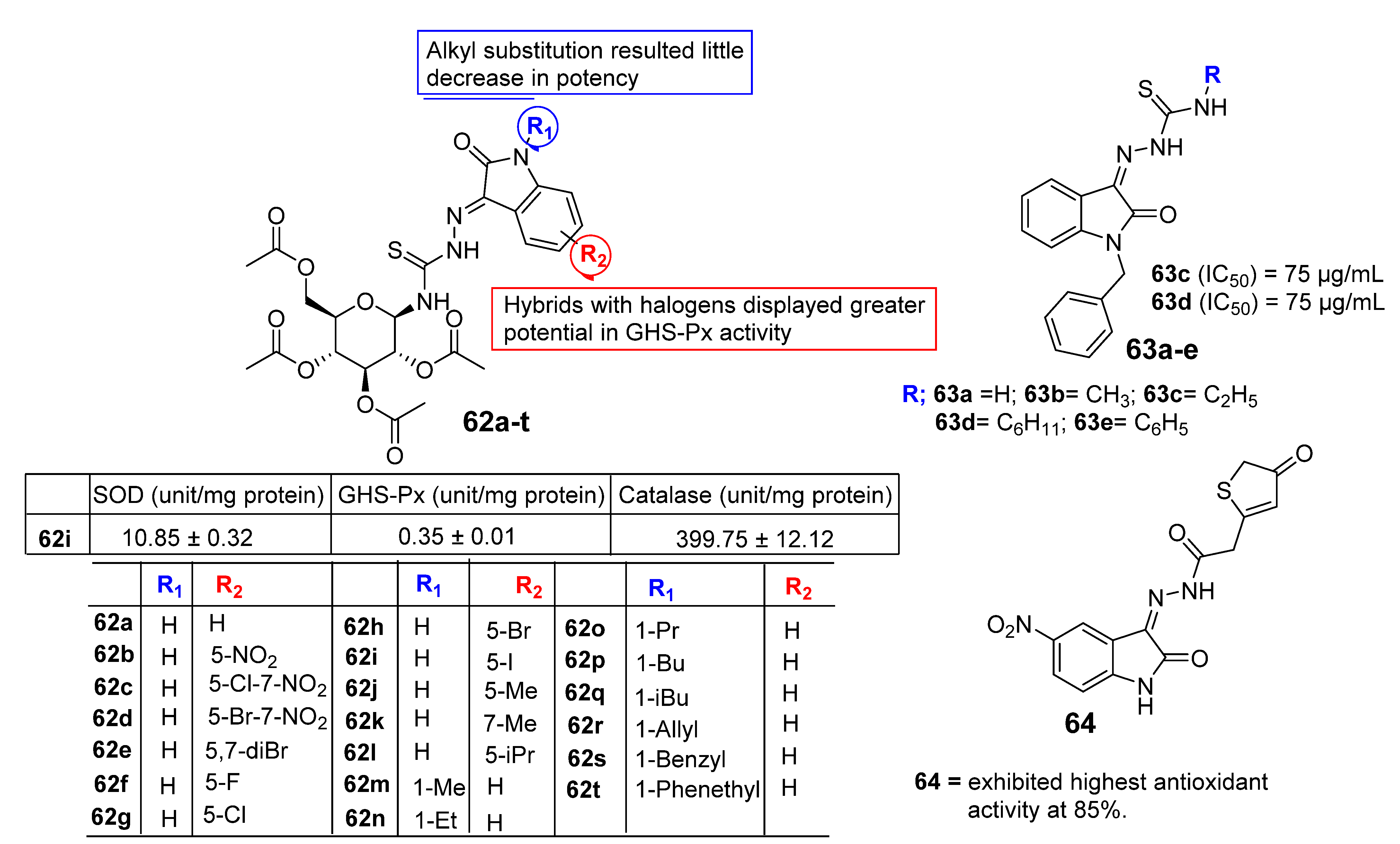
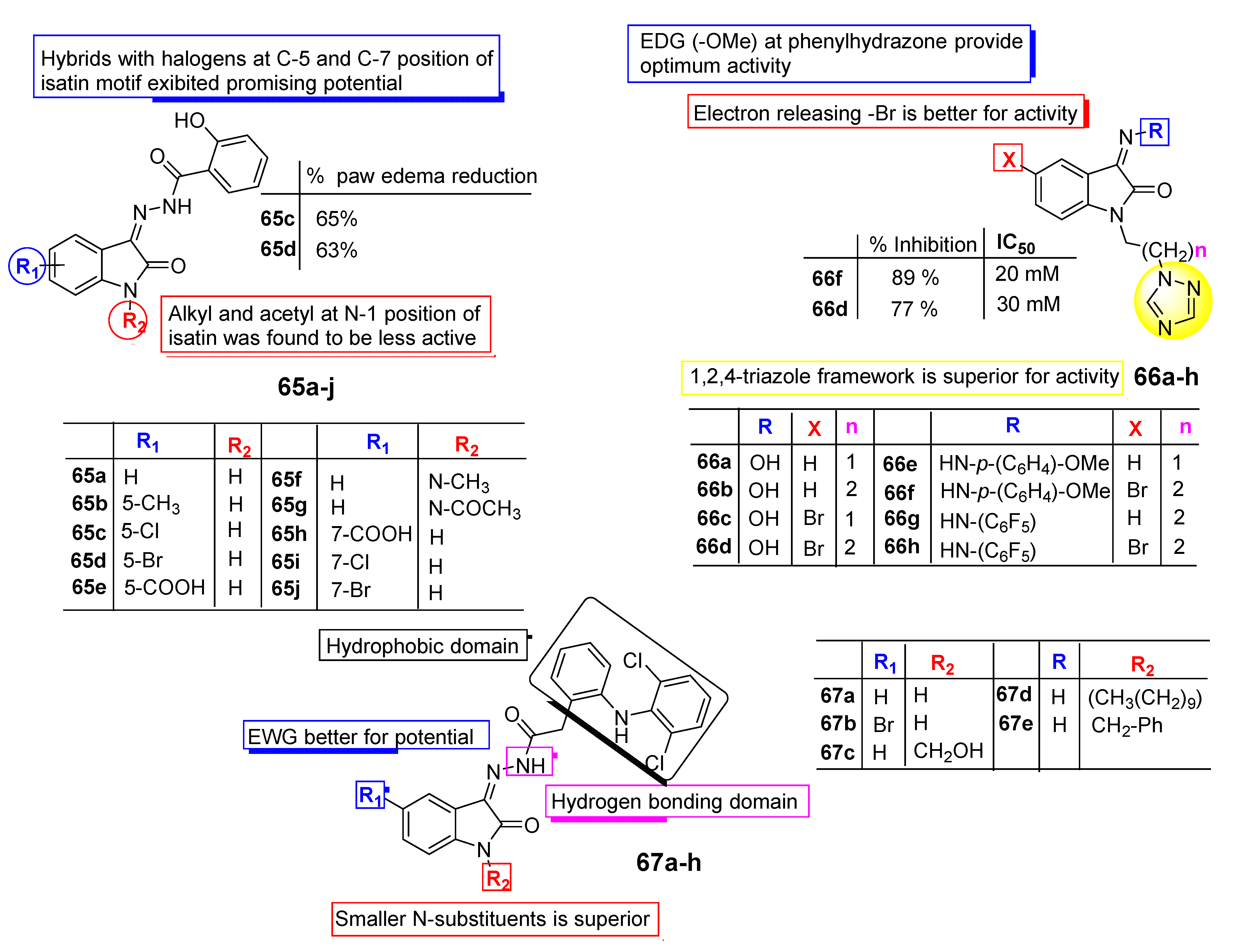
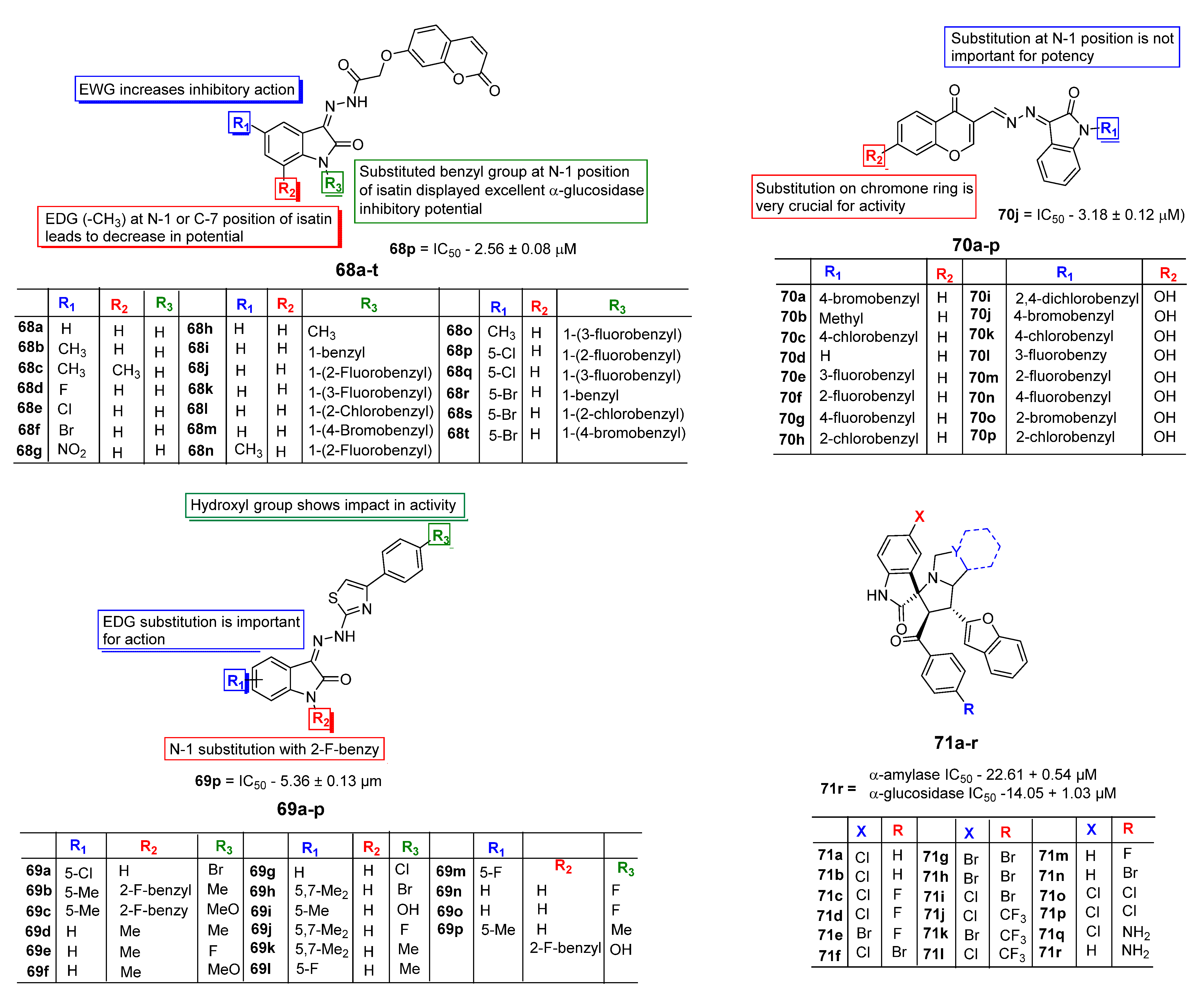
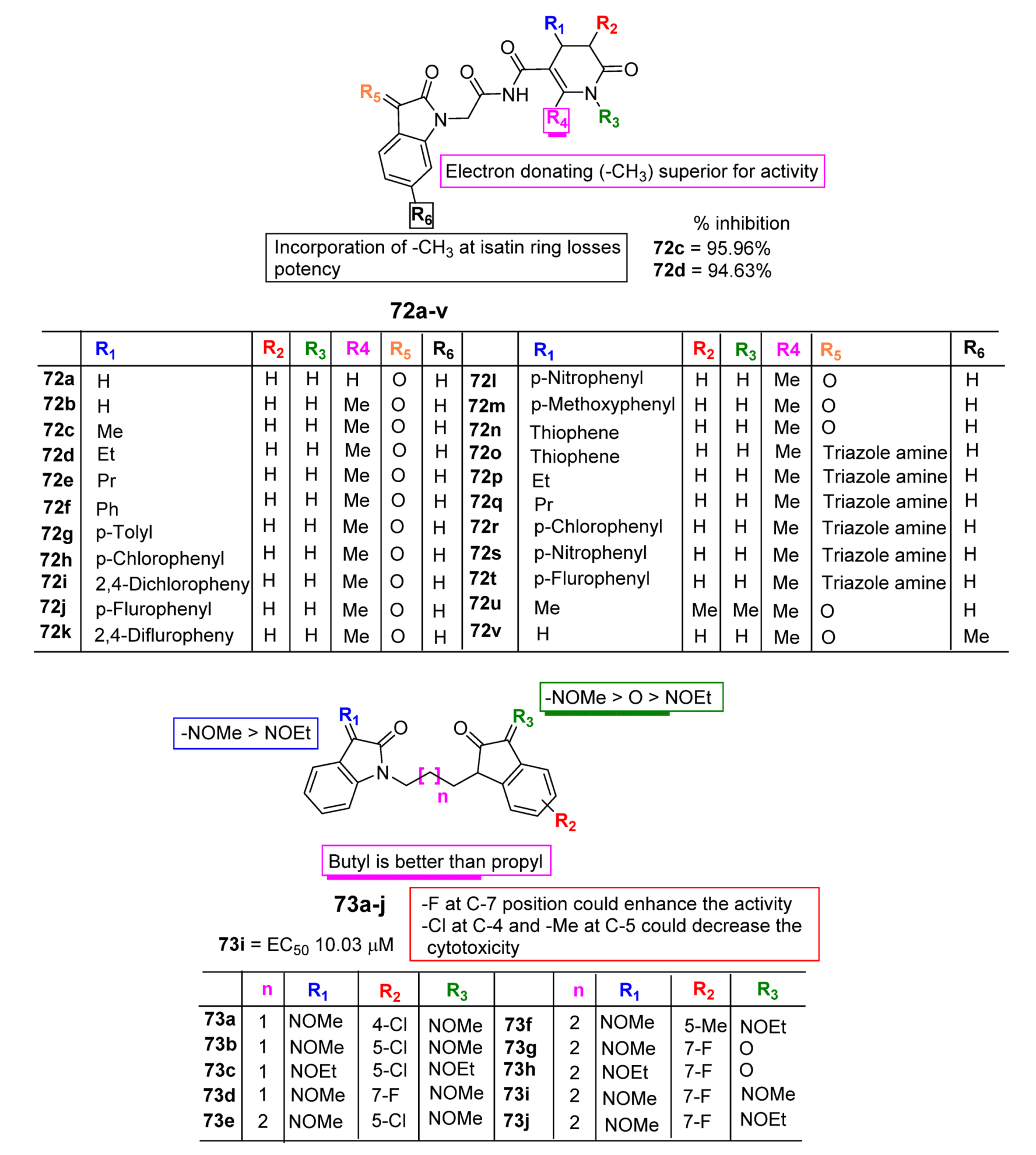
3. SAR Study for Promising Actions
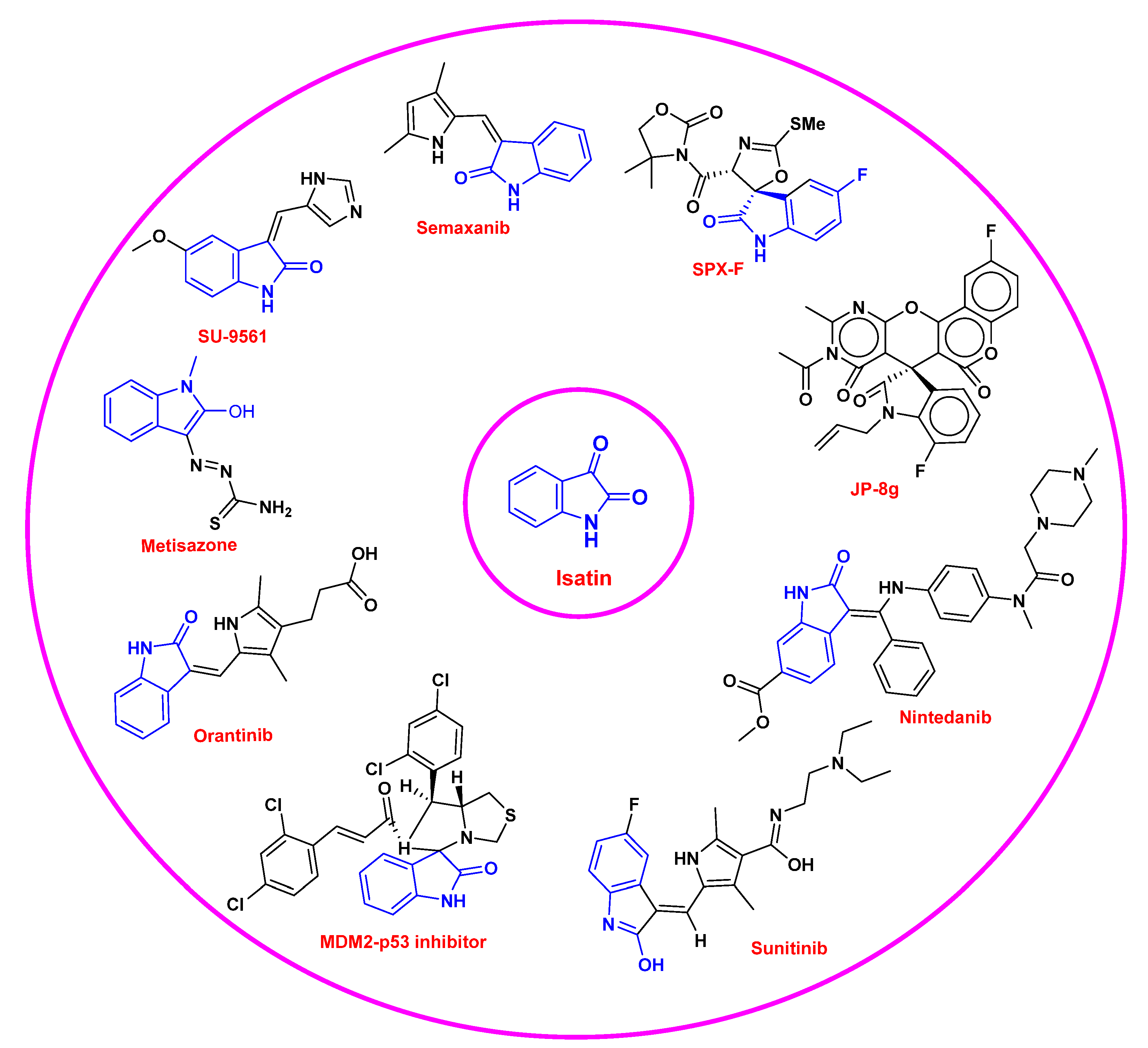
4. Clinical Developments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Shinde, S.D.; Ambhore, J.P.; Chaudhari, S.R.; Bari, S.B. Quinazoline: An update on current status against convulsions. J. Mol. Struct. 2022, 1248, 131384. [Google Scholar] [CrossRef]
- Varun; Sonam; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Med. Chem. Commun. 2019, 10, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.; Bhoi, M.; Prajapati, N.; Patel, D.; Francis, T. Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemistry Publication details, including instructions for authors and subscription information Review of Synthesis of Spiro Heterocyclic Compounds from Isatin. Synth. Commun. 2013, 447, 897–922. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Murakami, T.; Kishi, A.; Sakurama, T.; Matsuda, H.; Nomura, M.; Matsuda, H.; Kubo, M. Novel Indole S, O-Bisdesmoside, Calanthoside, The Precursor Glycoside of Tryptanthrin, Indirubin, and Isatin, with Increasing Skin Blood Flow Promoting Effects, from Two Calanthe Species (Orchidaceae). Chem. Pharm. Bull. 1998, 46, 886–888. [Google Scholar] [CrossRef]
- Bergman, J.; Lindström, J.-O.; Tilstam, U. The structure and properties of some indolic constituents in Couroupita guianensis aubl. Tetrahedron 1985, 41, 2879–2881. [Google Scholar] [CrossRef]
- Khan, F.; Maalik, A. Advances in Pharmacology of Isatin and its Derivatives: A Review. Trop. J. Pharm. Res. 2015, 14, 1937. [Google Scholar] [CrossRef] [Green Version]
- Chiyanzu, I.; Hansell, E.; Gut, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and evaluation of isatins and thiosemicarbazone derivatives against cruzain, falcipain-2 and rhodesain. Bioorg. Med. Chem. Lett. 2003, 13, 3527–3530. [Google Scholar] [CrossRef]
- Ke, S.; Shi, L.; Yang, Z. Discovery of novel isatin–dehydroepiandrosterone conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 25, 4628–4631. [Google Scholar] [CrossRef]
- Pawar, V.S.; Lokwani, D.K.; Bhandari, S.V.; Bothara, K.G.; Chitre, T.S.; Devale, T.L.; Modhave, N.S.; Parikh, J.K. Design, docking study and ADME prediction of Isatin derivatives as anti-HIV agents. Med. Chem. Res. 2011, 20, 370–380. [Google Scholar] [CrossRef]
- Abbas, S.Y.; Farag, A.A.; Ammar, Y.A.; Atrees, A.A.; Mohamed, A.F.; El-Henawy, A.A. Synthesis, characterization, and antiviral activity of novel fluorinated isatin derivatives. Mon. Fur Chem. 2013, 144, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xia, J.; Lei, D.; Li, X.; Yao, Q.; Gao, J. Synthesis, in vitro and in vivo antitumor activity of symmetrical bis-Schiff base derivatives of isatin. Eur. J. Med. Chem. 2014, 74, 742–750. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Sheikh, J.; El Mounsi, I.; Juneja, H.; Hadda, T. Ben Computational evaluation and experimental in vitro antibacterial, antifungal and antiviral activity of bis-Schiff bases of isatin and its derivatives. Med. Chem. Res. 2013, 22, 1203–1211. [Google Scholar] [CrossRef]
- Raj, R.; Biot, C.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Gut, J.; Rosenthal, P.J.; Forge, D.; Kumar, V. 7-Chloroquinoline–isatin Conjugates: Antimalarial, Antitubercular, and Cytotoxic Evaluation. Chem. Biol. Drug Des. 2014, 83, 622–629. [Google Scholar] [CrossRef]
- Venkateshwarlu, D.; Kulandaivelu, U.; Sheshagiri, S.; Jupalli, V. Evaluation of Antioxidant, Antimicrobial and Anticancer activity of Thiazole Tagged Isatin Hydrazones. J. Pharm. Chem. 2016, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Rajanarendar, E.; Ramakrishna, S.; Govardhan Reddy, K.; Nagaraju, D.; Reddy, Y.N. A facile synthesis, anti-inflammatory and analgesic activity of isoxazolyl-2,3-dihydrospiro[benzo[f]isoindole-1,3′-indoline]-2′,4,9-triones. Bioorg. Med. Chem. Lett. 2013, 23, 3954–3958. [Google Scholar] [CrossRef]
- El-Faham, A.; Hozzein, W.N.; Wadaan, M.A.M.; Khattab, S.N.; Ghabbour, H.A.; Fun, H.-K.; Siddiqui, M.R. Microwave Synthesis, Characterization, and Antimicrobial Activity of Some Novel Isatin Derivatives. J. Chem. 2015, 2015, 716987. [Google Scholar] [CrossRef] [Green Version]
- Mondal, P.; Jana, S.; Balaji, A.; Ramakrishna, R.; Kanthal, L.K. Synthesis of Some New Isoxazoline Derivatives of Chalconised Indoline 2-one as a Potential Analgesic, Antibacterial and Anthelmimtic Agents. J. Young Pharm. 2012, 4, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Firke, S.; Cheke, R.; Ugale, V.; Khadse, S.; Gagarani, M.; Surana, S.B. Rationale design, synthesis, and pharmacological evaluation of isatin analogues as antiseizure agents. Lett. Drug Des. Discov. 2021, 18, 1. [Google Scholar] [CrossRef]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, S.; Gao, C.; Fan, J.; Zhao, F.; Lv, Z.-S.; Feng, L.-S. Isatin hybrids and their anti-tuberculosis activity. Chin. Chem. Lett. 2017, 28, 159–167. [Google Scholar] [CrossRef]
- Ding, Z.; Zhou, M.; Zeng, C. Recent advances in isatin hybrids as potential anticancer agents. Arch. Pharm. 2020, 353, 1900367. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.-J.; Lv, Z.-S.; Gao, F.; Wang, Y.; Zhang, F.; Bai, L.; Deng, J.-L. Fluoroquinolone-isatin hybrids and their biological activities. Eur. J. Med. Chem. 2019, 162, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Firke, S.D.; Patil, R.R.; Bari, S.B. Isatin: New hope against convulsion. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 76–101. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Abou-seri, S.M.; Ismail, N.S.M.; Elaasser, M.M.; Aly, M.H.; Abdel-Aziz, H.A. Bis-isatin hydrazones with novel linkers: Synthesis and biological evaluation as cytotoxic agents. Eur. J. Med. Chem. 2016, 108, 415–422. [Google Scholar] [CrossRef]
- Teng, Y.-O.; Zhao, H.-Y.; Wang, J.; Liu, H.; Gao, M.-L.; Zhou, Y.; Han, K.-L.; Fan, Z.-C.; Zhang, Y.-M.; Sun, H.; et al. Synthesis and anti-cancer activity evaluation of 5-(2-carboxyethenyl)-isatin derivatives. Eur. J. Med. Chem. 2016, 112, 145–156. [Google Scholar] [CrossRef]
- Yu, B.; Wang, S.-Q.; Qi, P.-P.; Yang, D.-X.; Tang, K.; Liu, H.-M. Design and synthesis of isatin/triazole conjugates that induce apoptosis and inhibit migration of MGC-803 cells. Eur. J. Med. Chem. 2016, 124, 350–360. [Google Scholar] [CrossRef]
- Ammar, Y.A.; Fayed, E.A.; Bayoumi, A.H.; Ezz, R.R.; Alsaid, M.S.; Soliman, A.M.; Ghorab, M.M. New chalcones bearing isatin scaffold: Synthesis, molecular modeling and biological evaluation as anticancer agents. Res. Chem. Intermed. 2017, 43, 6765–6786. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Keeton, A.B.; Piazza, G.A.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S.; Attia, M.I. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des. Devel. Ther. 2017, 11, 2333–2346. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N. Isatin-β-thiocarbohydrazones: Microwave-assisted synthesis, antitumor activity and structure-activity relationship. Eur. J. Med. Chem. 2017, 128, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Gangarapu, K.; Thumma, G.; Manda, S.; Jallapally, A.; Jarapula, R.; Rekulapally, S. Design, synthesis and molecular docking of novel structural hybrids of substituted isatin based pyrazoline and thiadiazoline as antitumor agents. Med. Chem. Res. 2017, 26, 819–829. [Google Scholar] [CrossRef]
- Li, W.; Zhao, S.-J.; Gao, F.; Lv, Z.-S.; Tu, J.-Y.; Xu, Z. Synthesis and in Vitro Anti-Tumor, Anti-Mycobacterial and Anti-HIV Activities of Diethylene-Glycol-Tethered Bis-Isatin Derivatives. ChemistrySelect 2018, 3, 10250–10254. [Google Scholar] [CrossRef]
- Ammar, Y.A.; El-Sharief, A.M.S.; Belal, A.; Abbas, S.Y.; Mohamed, Y.A.; Mehany, A.B.M.; Ragab, A. Design, synthesis, antiproliferative activity, molecular docking and cell cycle analysis of some novel (morpholinosulfonyl) isatins with potential EGFR inhibitory activity. Eur. J. Med. Chem. 2018, 156, 918–932. [Google Scholar] [CrossRef]
- Zou, Y. Benzofuran-isatin conjugates as potent VEGFR-2 and cancer cell growth inhibitors. J. Heterocycl. Chem. 2020, 57, 510–516. [Google Scholar] [CrossRef]
- Alkahtani, H.M.; Alanazi, M.M.; Aleanizy, F.S.; Alqahtani, F.Y.; Alhoshani, A.; Alanazi, F.E.; Almehizia, A.A.; Abdalla, A.N.; Alanazi, M.G.; El-Azab, A.S.; et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019, 27, 682–693. [Google Scholar] [CrossRef]
- Xu, K.; Liu, Y.; Wang, R.; Cai, P.; Fang, Y. Design, Synthesis, and Anticancer Activities of Benzofuran–Isatin Hybrids Tethered by Pentylene and Hexylene. J. Heterocycl. Chem. 2019, 56, 2052–2055. [Google Scholar] [CrossRef]
- Jiang, D.; Zhang, G. Ciprofloxacin/Gatifloxacin-1,2,3-triazole-isatin Hybrids and Their in Vitro Anticancer Activity. J. Heterocycl. Chem. 2019, 56, 2966–2969. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Almomen, A.A.; Almutairi, M.S.; Keeton, A.B.; Piazza, G.A.; Attia, M.I. New Isatin-Indole Conjugates: Synthesis, Characterization, and a Plausible Mechanism of Their in vitro Antiproliferative Activity. Drug Des. Devel. Ther. 2020, 14, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Rajesh Kumar, M.; Violet Dhayabaran, V.; Sudhapriya, N.; Manikandan, A.; Gideon, D.A.; Annapoorani, S. p-TSA.H2O mediated one-pot, multi-component synthesis of isatin derived imidazoles as dual-purpose drugs against inflammation and cancer. Bioorg. Chem. 2020, 102, 104046. [Google Scholar] [CrossRef] [PubMed]
- Nazari, S.; Safari, F.; Mamaghani, M.B.; Bazgir, A. Synthesis and evaluation of in vitro cytotoxic effects of triazol/spiroindolinequinazolinedione, triazol/indolin-3-thiosemicarbazone and triazol/thiazol-indolin-2-one conjugates. DARU J. Pharm. Sci. 2020, 28, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Subhedar, D.D.; Khan, F.A.K.; Sangshetti, J.N.; Nawale, L.; Arkile, M.; Sarkar, D.; Shingate, B.B. Synthesis of Novel Triazole-incorporated Isatin Derivatives as Antifungal, Antitubercular, and Antioxidant Agents and Molecular Docking Study. J. Heterocycl. Chem. 2017, 54, 413–421. [Google Scholar] [CrossRef]
- Borad, M.A.; Bhoi, M.N.; Rathwa, S.K.; Vasava, M.S.; Patel, H.D.; Patel, C.N.; Pandya, H.A.; Pithawala, E.A.; Georrge, J.J. Microwave-Assisted ZrSiO2 Catalysed Synthesis, Characterization and Computational Study of Novel Spiro[Indole-Thiazolidines] Derivatives as Anti-tubercular Agents. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 411–418. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, S.; Song, X.; Qiang, M.; Lv, Z. Design, synthesis and in vitro anti-mycobacterial evaluation of gatifloxacin-1H-1,2,3-triazole-isatin hybrids. Bioorg. Med. Chem. Lett. 2017, 27, 3643–3646. [Google Scholar] [CrossRef]
- Hu, Y.-Q.; Meng, L.-D.; Qiang, M.; Song, X.-F. Design, Synthesis, and in Vitro Anti-mycobacterial Evaluation 1H-1,2,3-triazole-tethered Ciprofloxacin and Isatin Conjugates. J. Heterocycl. Chem. 2017, 54, 3725–3729. [Google Scholar] [CrossRef]
- Yan, X.; Lv, Z.; Wen, J.; Zhao, S.; Xu, Z. Synthesis and in vitro evaluation of novel substituted isatin-propylene-1H-1,2,3-triazole-4-methylene-moxifloxacin hybrids for their anti-mycobacterial activities. Eur. J. Med. Chem. 2018, 143, 899–904. [Google Scholar] [CrossRef]
- Xu, Z.; Song, X.-F.; Qiang, M.; Lv, Z.-S. 1H-1,2,3-triazole-tethered 8-OMe Ciprofloxacin and Isatin Hybrids: Design, Synthesis and in vitro Anti-mycobacterial Activities. J. Heterocycl. Chem. 2017, 54, 3735–3741. [Google Scholar] [CrossRef]
- Xu, Z.; Lv, Z.-S.; Song, X.-F.; Qiang, M. Ciprofloxacin-isatin-1H-1,2,3-triazole Hybrids: Design, Synthesis, and in vitro Anti-tubercular Activity against M. tuberculosis. J. Heterocycl. Chem. 2018, 55, 97–102. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, J.; Xu, Z.; Zhao, S. Design, synthesis and in vitro anti-mycobacterial activities of homonuclear and heteronuclear bis-isatin derivatives. Fitoterapia 2018, 127, 383–386. [Google Scholar] [CrossRef]
- Zhao, S.-Q.; Xu, Y.; Guan, J.; Zhao, S.; Zhang, G.-D.; Xu, Z. Tetraethylene Glycol Tethered Heteronuclear Bis-isatin Derivatives: Design, Synthesis, and in Vitro Anti-mycobacterial Activities. J. Heterocycl. Chem. 2018, 55, 2172–2177. [Google Scholar] [CrossRef]
- Gao, F.; Yang, H.; Lu, T.; Chen, Z.; Ma, L.; Xu, Z.; Schaffer, P.; Lu, G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur. J. Med. Chem. 2018, 159, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Zhang, G.; Zhang, D.; Wu, Y. Design, Synthesis, and in vitro Anti-mycobacterial Activities of Propylene-tethered Heteronuclear Bis-isatin Derivatives. J. Heterocycl. Chem. 2018, 55, 1504–1508. [Google Scholar] [CrossRef]
- Gao, T.; Zeng, Z.; Wang, G.; Sun, S.; Liu, Y. Synthesis of Ethylene Tethered Isatin-Coumarin Hybrids and Evaluation of Their in vitro Antimycobacterial Activities. J. Heterocycl. Chem. 2018, 55, 1484–1488. [Google Scholar] [CrossRef]
- Wang, H.-D.; Fan, Y.-L.; Zhou, J.; Xu, Y.; Guan, J. Design, Synthesis, and in Vitro Anti-mycobacterial Activities of Propylene-Tethered Gatifloxacin-Isatin Hybrids. J. Heterocycl. Chem. 2018, 55, 1991–1996. [Google Scholar] [CrossRef]
- Xu, Y.; Dang, R.; Guan, J.; Xu, Z.; Zhao, S.; Hu, Y. Isatin-(thio)semicarbazide/oxime-1H-1,2,3-triazole-coumarin Hybrids: Design, Synthesis, and in vitro Anti-mycobacterial Evaluation. J. Heterocycl. Chem. 2018, 55, 1069–1073. [Google Scholar] [CrossRef]
- Jialun, D.; Liu, X.-C.; Guangwei, C.; Gang, Z.; Li, H.; Li, Q.; Ziyong, L.; Xu, Z. Heteronuclear 5-Fluoroisatin Dimers: Design, Synthesis, and Evaluation of Their in Vitro Anti-mycobacterial Activities: Heteronuclear 5-Fluoroisatin Dimers. J. Heterocycl. Chem. 2018, 55, 1509–1513. [Google Scholar]
- Wang, R.; Yin, X.; Zhang, Y.; Yan, W. Design, synthesis and antimicrobial evaluation of propylene-tethered ciprofloxacin-isatin hybrids. Eur. J. Med. Chem. 2018, 156, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, T.; Gao, M.; Zhang, X.; Liu, Z.; Zhao, S.; Lv, Z.; Xiao, J. Benzofuran-isatin-imine hybrids tethered via different length alkyl linkers: Design, synthesis and in vitro evaluation of anti-tubercular and anti-bacterial activities as well as cytotoxicity. Eur. J. Med. Chem. 2019, 165, 323–331. [Google Scholar] [CrossRef]
- Gao, F.; Ye, L.; Wang, Y.; Kong, F.; Zhao, S.; Xiao, J.; Huang, G. Benzofuran-isatin hybrids and their in vitro anti-mycobacterial activities against multi-drug resistant Mycobacterium tuberculosis. Eur. J. Med. Chem. 2019, 183, 111678. [Google Scholar] [CrossRef]
- Gao, F.; Chen, Z.; Ma, L.; Fan, Y.; Chen, L.; Lu, G. Synthesis and biological evaluation of moxifloxacin-acetyl-1,2,3-1H-triazole-methylene-isatin hybrids as potential anti-tubercular agents against both drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. Eur. J. Med. Chem. 2019, 180, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, H.; Ma, T.; Xue, H.; Miao, Z.; Chen, L.; Shi, X. Ciprofloxacin-1,2,3-triazole-isatin hybrids tethered via amide: Design, synthesis, and in vitro anti-mycobacterial activity evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 2635–2637. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, G.-Q.; Peng, Y.-H.; Tang, X.-Q.; Hu, G.-W. Design, synthesis, and in vitro antimycobacterial activities of butylene tethered 7-fluoroisatin-isatin scaffolds. J. Heterocycl. Chem. 2019, 56, 3423–3428. [Google Scholar] [CrossRef]
- Ding, Z.; Hou, P.; Liu, B. Gatifloxacin-1,2,3-triazole-isatin hybrids and their antimycobacterial activities. Arch. Pharm. 2019, 352, e1900135. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, Z.M.; Eldehna, W.M.; Abdel-Aziz, M.M.; El Hassab, M.A.; Elkaeed, E.B.; Al-Warhi, T.; Abdel-Aziz, H.A.; Abou-Seri, S.M.; Mohammed, E.R. Development of novel isatin-nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing-bacteria. J. Enzym. Inhib. Med. Chem. 2021, 36, 384–393. [Google Scholar] [CrossRef]
- Lian, Z.-M.; Sun, J.; Zhu, H.-L. Design, synthesis and antibacterial activity of isatin derivatives as FtsZ inhibitors. J. Mol. Struct. 2016, 1117, 8–16. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Zakaria, A.S.; Attia, M.I. Synthesis, Spectroscopic Characterization and Antimicrobial Potential of Certain New Isatin-Indole Molecular Hybrids. Molecules 2017, 22, 1958. [Google Scholar] [CrossRef] [Green Version]
- Kenchappa, R.; Bodke, Y.D.; Telkar, S.; Nagaraja, O. Synthesis and antimicrobial activity of fused isatin and diazepine derivatives derived from 2-acetyl benzofuran. Russ. J. Gen. Chem. 2017, 87, 2027–2038. [Google Scholar] [CrossRef]
- Almutairi, M.; Zakaria, A.; Primsa, I.P.; Al-wabli, R.; Joe, I.H.; Attia, M. Synthesis, spectroscopic investigations, DFT studies, molecular docking and antimicrobial potential of certain new indole-isatin molecular hybrids: Experimental and theoretical approaches. J. Mol. Struct. 2017, 1153, 335–345. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Zaripova, I.F.; Voloshina, A.D.; Strobykina, A.S.; Kulik, N.V.; Bukharov, S.V.; Voronina, J.K.; Khamatgalimov, A.R.; Mironov, V.F. Synthesis and antimicrobial activity evaluation of some novel water-soluble isatin-3-acylhydrazones. Mon. Für Chem.-Chem. Mon. 2018, 149, 111–117. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Singh, A.; Kalra, P.; Rani, S.; Singh, K.; Maurya, I.K.; Mandal, R.S. Molecular Design, Synthesis, Computational Screening, Antimicrobial Evaluation and Molecular Docking Study of Acetylinic Isatin Hybrids. ChemistrySelect 2018, 3, 1942–1952. [Google Scholar] [CrossRef]
- Gomaa, M.A.-M.; Hassan, D.K. Synthesis, characterization, and antimicrobial activity of some new N-aryl-N′-(2-oxoindolin-3-ylidene)-benzohydrazonamides. Arch. Pharm. 2019, 352, e1900209. [Google Scholar] [CrossRef]
- Gao, F.; Ye, L.; Kong, F.; Huang, G.; Xiao, J. Design, synthesis and antibacterial activity evaluation of moxifloxacin-amide-1,2,3-triazole-isatin hybrids. Bioorg. Chem. 2019, 91, 103162. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, K.; Bhagat, J.; Gupta, M.K.; Singh, J.V.; Gulati, H.K.; Singh, A.; Kaur, K.; Kaur, G.; Sharma, S.; Rana, A.; et al. Design, Synthesis, Antimicrobial Evaluation, and Molecular Modeling Studies of Novel Indolinedione–Coumarin Molecular Hybrids. ACS Omega 2019, 4, 8720–8730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahari, K.; Sundararajan, R. Design and synthesis of novel isatin derivatives as potent analgesic, anti-inflammatory and antimicrobial agents. J. Chem. Sci. 2020, 132, 94. [Google Scholar] [CrossRef]
- Mangasuli, S. Synthesis of novel Isatin-Dithiocarbamate hybrids: An approach to Microwave and potent antimicrobial agents. Chem. Data Collect. 2020, 29, 100515. [Google Scholar] [CrossRef]
- Rawat, P.; Verma, S.M. Design and synthesis of chroman derivatives with dual anti-breast cancer and antiepileptic activities. Drug Des. Devel. Ther. 2016, 10, 2779–2788. [Google Scholar] [CrossRef] [Green Version]
- Rahmani-Khajouei, M.; Mohammadi-Farani, A.; Mirzaei, D.; Aliabadi, A. Isatin-based anticonvulsant agents: Synthesis and antiseizure evaluation in mice. J. Rep. Pharm. Sci. 2017, 6, 13–22. [Google Scholar] [CrossRef]
- Divar, M.; Yeganeh, Y.; Jamshidzadeh, A.; Reza Heidari, S.K. Anticonvulsant activity of some semicarbazone and thiosemicarbazone derivatives of isatin on PTZ induced seizure. J. Innov. Appl. Pharm. Sci. 2017, 2, 4–14. [Google Scholar]
- Kiranmai, M. Synthesis and Evaluation of Some Novel N, N-Dialkylaminoalkoxy-2-Oxo-Indole-3-Ylidene Benzohydrazides as Anticonvulsant Agents. IOSR J. Pharm. Biol. Sci. 2017, 12, 84–93. [Google Scholar] [CrossRef]
- Khajouei, M.R.; Mohammadi-Farani, A.; Moradi, A.; Aliabadi, A. Synthesis and evaluation of anticonvulsant activity of (Z)-4-(2-oxoindolin-3-ylideneamino)-N-phenylbenzamide derivatives in mice. Res. Pharm. Sci. 2018, 13, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Nath, R.; Shahar Yar, M.; Pathania, S.; Grover, G.; Debnath, B.; Akhtar, M.J. Synthesis and anticonvulsant evaluation of indoline derivatives of functionalized aryloxadiazole amine and benzothiazole acetamide. J. Mol. Struct. 2021, 1228, 129742. [Google Scholar] [CrossRef]
- Thanh, N.D.; Giang, N.T.K.; Quyen, T.H.; Huong, D.T.; Toan, V.N. Synthesis and evaluation of in vivo antioxidant, in vitro antibacterial, MRSA and antifungal activity of novel substituted isatin N-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazones. Eur. J. Med. Chem. 2016, 123, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, J.; Subhashree, G.R.; Saranya, S.; Gomathi, K.; Karvembu, R.; Gayathri, D. Isatin based thiosemicarbazone derivatives as potential bioactive agents: Anti-oxidant and molecular docking studies. J. Mol. Struct. 2016, 1110, 185–195. [Google Scholar] [CrossRef]
- El-Serwy, W.; Mohamed, N.; El-Serwy, W.; Kassem, E.; Shalaby, A. Synthesis, antioxidant, anticoagulant, and fibrinolytic activities of new isatin derivative. Egypt. Pharm. J. 2020, 19, 113. [Google Scholar] [CrossRef]
- Jarapula, R.; Gangarapu, K.; Manda, S.; Rekulapally, S. Synthesis, in Vivo Anti-Inflammatory Activity, and Molecular Docking Studies of New Isatin Derivatives. Int. J. Med. Chem. 2016, 2016, 2181027. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.K.; Balwani, S.; Mathur, D.; Malhotra, S.; Singh, B.K.; Prasad, A.K.; Len, C.; Van der Eycken, E.V.; Ghosh, B.; Richards, N.G.J.; et al. Synthesis and anti-inflammatory activity evaluation of novel triazolyl-isatin hybrids. J. Enzym. Inhib. Med. Chem. 2016, 31, 1520–1526. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.M.; Elsaman, T.; Al-Nour, M.Y. Synthesis, Anti-Inflammatory Activity, and in Silico Study of Novel Diclofenac and Isatin Conjugates. Int. J. Med. Chem. 2018, 2018, 9139786. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. Synthesis, in vitro evaluation and molecular docking studies of novel coumarin-isatin derivatives as α-glucosidase inhibitors. Chem. Biol. Drug Des. 2017, 89, 456–463. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, G.; Wang, J.; Chen, M.; Peng, Y.; Li, L.; Deng, B.; Chen, S.; Li, W. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors. Molecules 2017, 22, 659. [Google Scholar] [CrossRef]
- Wang, G.; Chen, M.; Qiu, J.; Xie, Z.; Cao, A. Synthesis, in vitro α-glucosidase inhibitory activity and docking studies of novel chromone-isatin derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Altowyan, M.S.; Barakat, A.; Al-Majid, A.M.; Al-Ghulikah, H.A. Spiroindolone Analogues as Potential Hypoglycemic with Dual Inhibitory Activity on α-Amylase and α-Glucosidase. Molecules 2019, 24, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devale, T.L.; Parikh, J.; Miniyar, P.; Sharma, P.; Shrivastava, B.; Murumkar, P. Dihydropyrimidinone-isatin hybrids as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Chem. 2017, 70, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, S.; Xu, Z.; Liu, Y. Investigation of the Anti-human Immunodeficiency Virus Activity of Heteronuclear Bis-isatin Scaffolds Tethered through Propyl and Butyl. J. Heterocycl. Chem. 2019, 56, 2975–2979. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 15 December 2021).
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
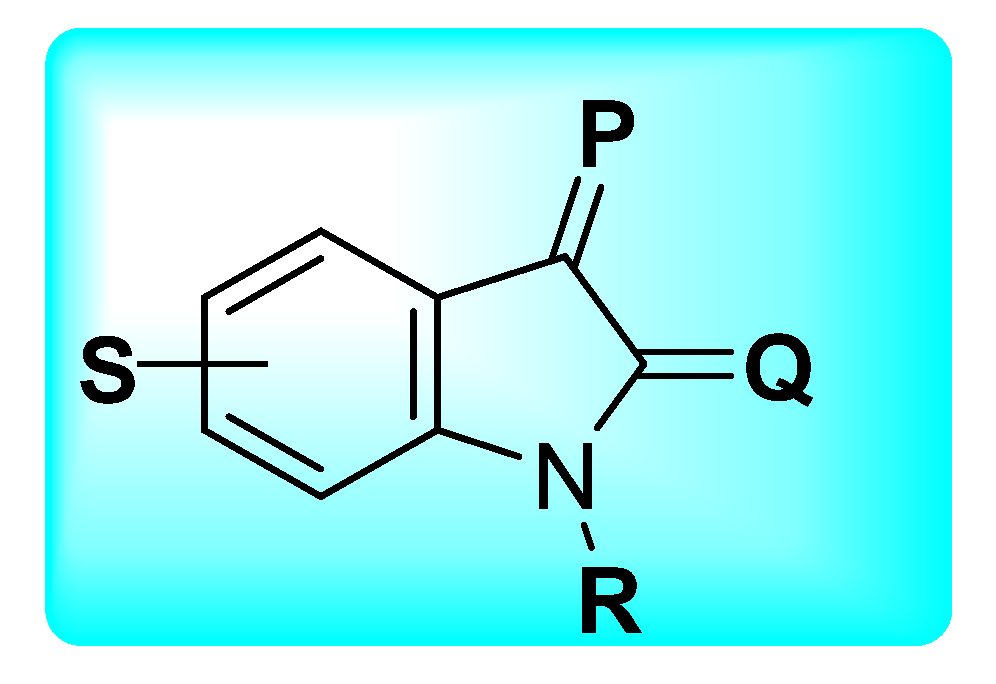
| Activities | P | Q | R | S |
|---|---|---|---|---|
| Anti-Cancer | Carbonyl species to form hydrazone, imines, or hydrazide resulting in potent CDK and Kinase inhibition. Addition of Imidazole and pyrrole leads to potent inhibition of receptor tyrosine kinase (RTK). | Hybrids with styrenes with EDG antagonize tyrosine kinase. Dimers enhance inhibition of tyrosine kinase CDK1 and 2. | Aromatic or aliphatic substitution shows courtesy towards microtubule destabilization. Para or meta substitution good for potency compared to ortho. | Halogen species favor antitumor potential |
| Anti-TB | Monosubstituted bis-isatin hybrids were more active than the bis-substituted and unsubstituted hybrids (NNHCSNH2 > O > NOMe > NOEt > NOH). | Carbonyl function is important for potency. Unsubstituted carbonyl function Improve the potential | C-1 positions of isatin moiety are deemed as the most favorable sites for hybridization. Incorporation alkyl chain linker, triazole linker displayed potent anti-TB action. | Electron withdrawing substitution at C-5 position of isatin resulted in excellent anti-TB potency. |
| Anti-Microbial | Schiff bases, thio-semicarbazones, semicarbazones, substituted oximes, hydrazones, etc., good for promising antimicrobial action. | Unsubstituted Q position favors noncovalent inactions at binding site. | Unsubstituted or alkyl chain linker, triazole linker resulted in good activity. | Aryl ring substituted with EWGs exhibited excellent antimicrobial action, occasionally EDGs are also good. |
| Anti-Convulsant | Schiff bases, thiosemicarbazones, semicarbazones with substituted aryl ring good for anticonvulsant potential. | Free to bind with receptor cavity. | Substitution at R eliminated the anticonvulsant action. | Substitution of small EWGs exhibited potential activity. |
| Anti-Oxidant | Imine, hydrazone, spiro, oxime oxindole substitution favors the antioxidant action. | Hydrogen bonding domain. | N-alkylation, aryl, acyl unfavorable for antioxidant potential. | EWGs such as halogens, -NO2 atC-5 increase significant enhancement in activity. |
| Anti-Inflammatory | Incorporation of EDGs substituted phenylhydrazone at C-3 position displayed added anti-inflammatory activity. | Hydrogen bonding domain. | Smaller N-substitution favors the anti-inflammatory potential. | EWGs at C-5 position of isatin motif favorable for anti-inflammatory action compared to EDGs. |
| Anti-Diabetic | Schiff bases, semicarbazones, and hydrazone link EDGs-substituted aryl, phenyl chromine ring promising for inhibitory action. | Hydrogen bonding domain. | N-substitution favors the anti-diabetic potential. | EWGs favor the inhibitory potential when compared to EDGs. |
| Anti-HIV | Schiff bases, semicarbazones, and hydrazone oximes could boost the potency. | No substitution is preferred. | N-substitution such as alkyl linker, triazole could improve anti-HIV potential. | EWGs at C-5 position could enhance anti-HIV profile. |
| Name of Drug | Year | FDA Approved Clinical Indications |
|---|---|---|
| Sunitinib | 2006 | Gastrointestinal stromal tumors and advanced renal cell carcinoma |
| 2011 | Pancreatic cancer | |
| 2017 | Adjuvant agent for recurrent renal carcinoma | |
| 2019 | Phase 2 in metastatic pancreatic neuroendocrine tumor | |
| 2020 | Metastatic renal cell carcinoma | |
| Toceranib | 2009 | Canine mast cell tumor |
| Nintedanib | 2018 | Phase 3 completed for refractory metastatic colorectal cancer |
| 2018 | Phase 3 completed for combination with Paclitaxel and Carboplatin for use in ovarian cancer (first line therapy) | |
| 2018 | Phase 3 completed for combination with Docetaxel for use in non-small cell lung cancer | |
| 2019 | Phase 1 completed for combination with Letrozole for breast cancer in postmenopausal women | |
| 2019 | Phase 2 completed for recurrent or metastatic breast cancer | |
| 2019 | Phase 2 terminated for metastatic HER2-negative inflammatory breast cancer | |
| 2019 | Phase 2 completed for advanced ovarian cancer | |
| Orantinib | 2011 | Phase1/2 completed for use in advanced hepatocellular carcinoma |
| 2017 | Phase 3 in hepatocellular carcinoma | |
| Semaxinib | 2003 | Phase 2 completed for use in persistent and recurrent cervical cancer |
| 2004 | Phase 3 completed for use as combination with 5-Fluorouracil, Leucovorin, and Irinotecan in metastatic colorectal cancer | |
| 2009 | Phase 2 completed for use in advanced/recurrent head and neck cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheke, R.S.; Patil, V.M.; Firke, S.D.; Ambhore, J.P.; Ansari, I.A.; Patel, H.M.; Shinde, S.D.; Pasupuleti, V.R.; Hassan, M.I.; Adnan, M.; et al. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals 2022, 15, 272. https://doi.org/10.3390/ph15030272
Cheke RS, Patil VM, Firke SD, Ambhore JP, Ansari IA, Patel HM, Shinde SD, Pasupuleti VR, Hassan MI, Adnan M, et al. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals. 2022; 15(3):272. https://doi.org/10.3390/ph15030272
Chicago/Turabian StyleCheke, Rameshwar S., Vaishali M. Patil, Sandip D. Firke, Jaya P. Ambhore, Iqrar A. Ansari, Harun M. Patel, Sachin D. Shinde, Visweswara Rao Pasupuleti, Md Imtaiyaz Hassan, Mohd Adnan, and et al. 2022. "Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery" Pharmaceuticals 15, no. 3: 272. https://doi.org/10.3390/ph15030272