
A Selective Inhibitor of Cardiac Troponin I Phosphorylation by Delta Protein Kinase C (δPKC) as a Treatment for Ischemia-Reperfusion Injury


 , , and
, , and
Abstract
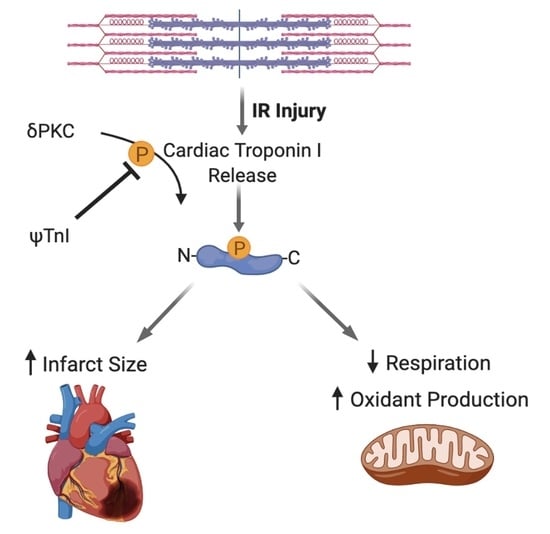
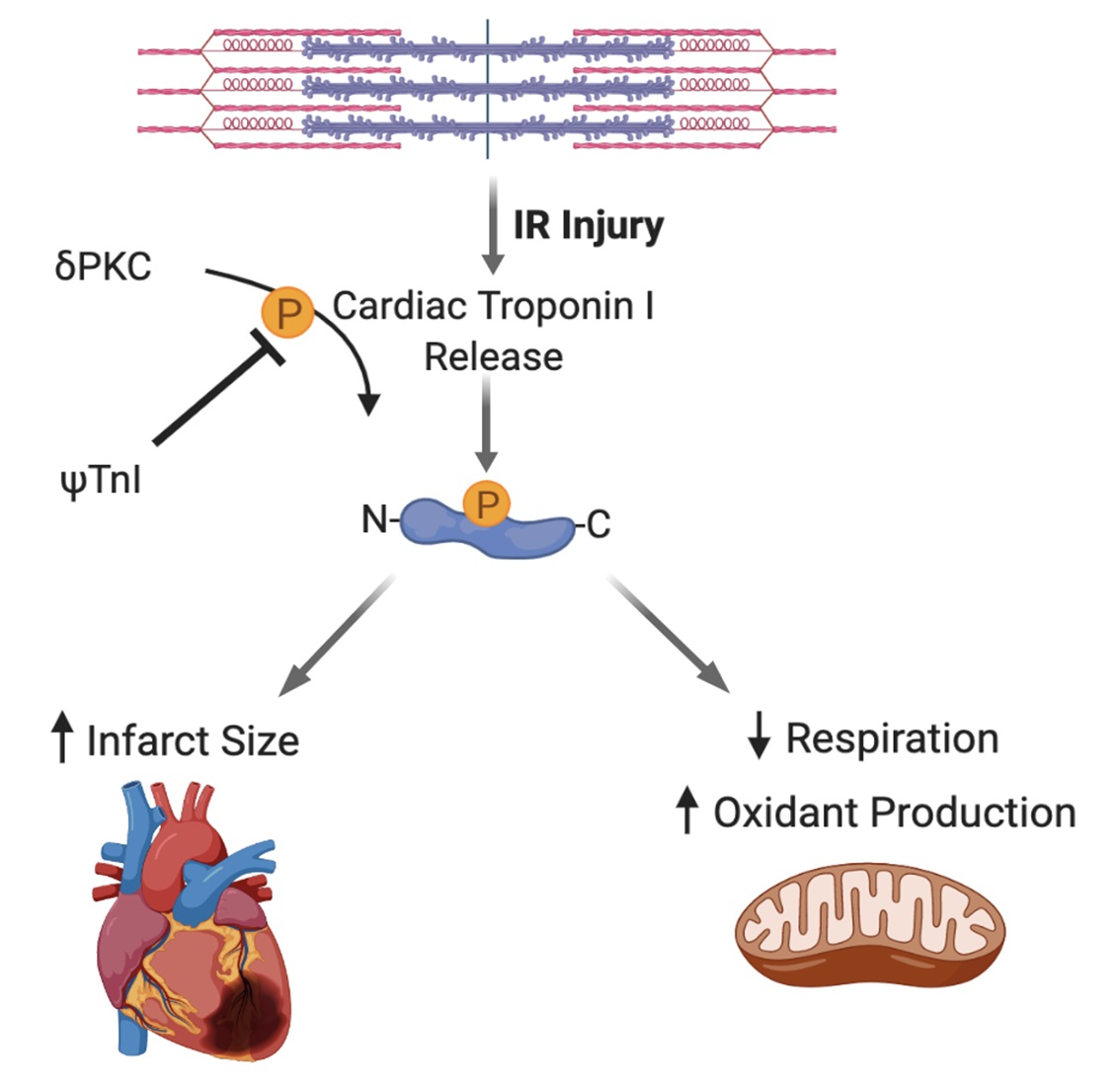
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
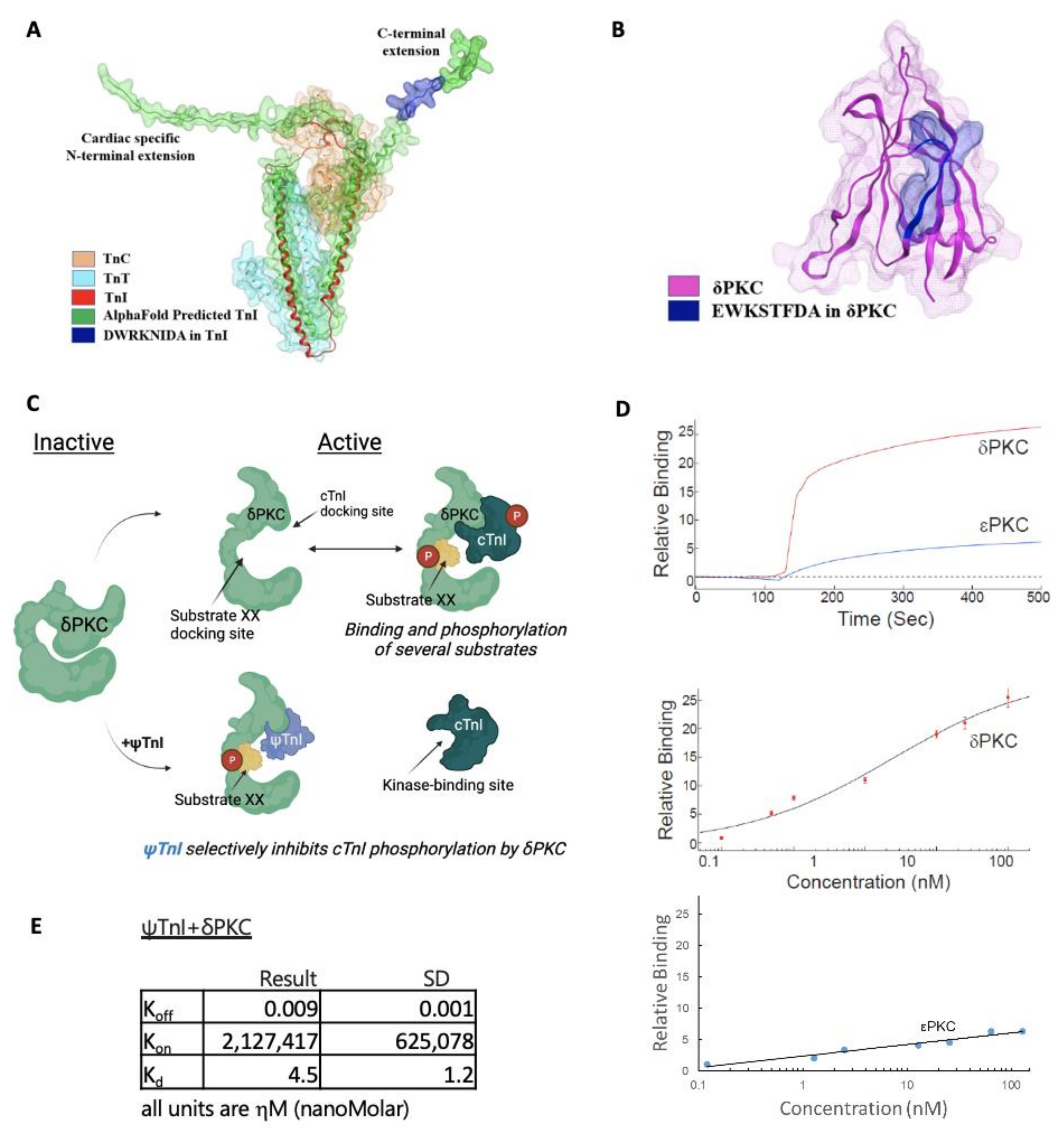
2.1. Design of a Selective Inhibitor of cTnI’s Interaction with δPKC
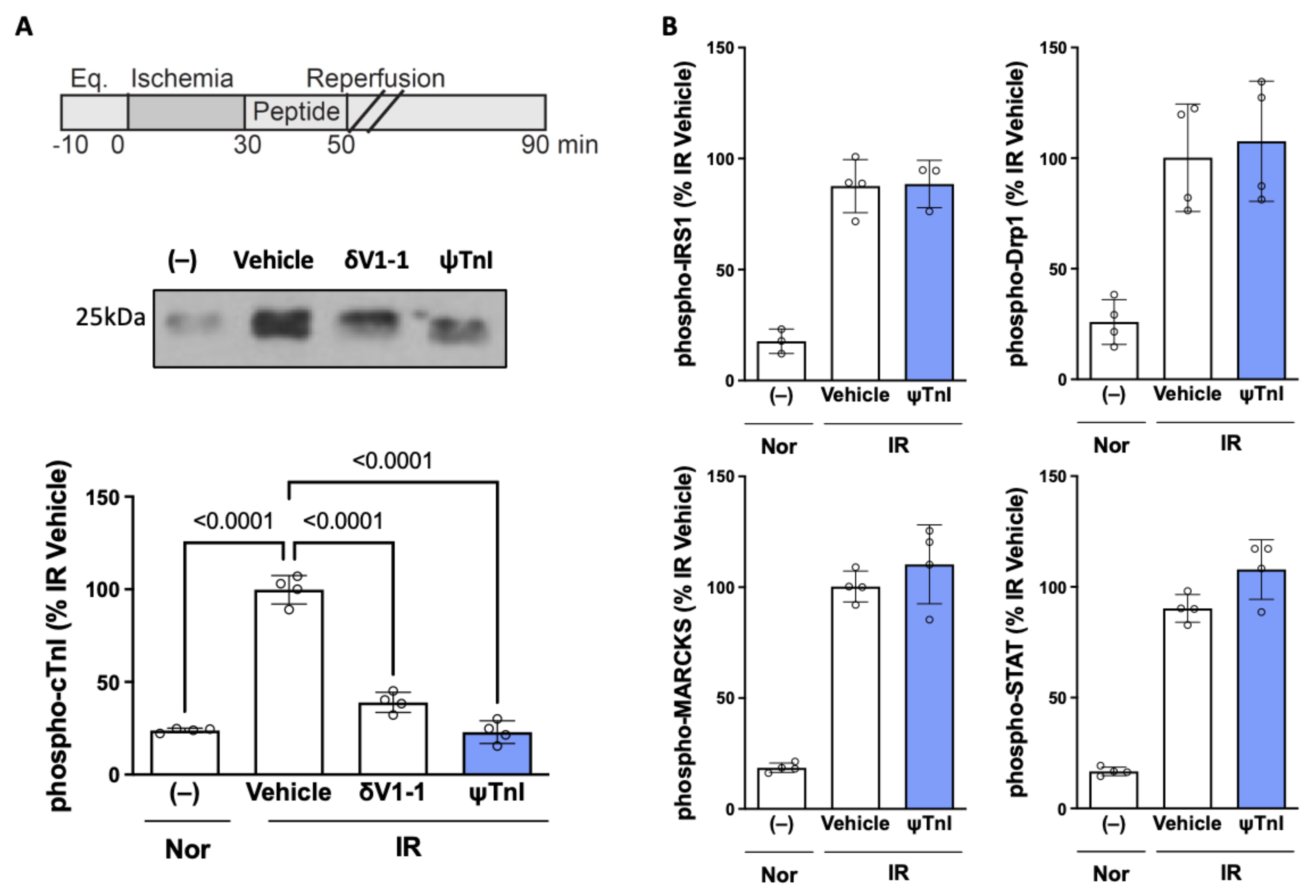
2.2. Selective Inhibition of cTnI Phosphorylation by δPKC Prevents Ex Vivo IR-Induced Myocardial Injury
2.3. Inhibition of cTnI Phosphorylation by δPKC Attenuates Ex Vivo IR-Induced Mitochondrial Dysfunction
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Peptide Synthesis
4.3. Peptide Binding to δPKC and εPKC in Vitro
4.4. Cardiac Ischemia Reperfusion—Ex Vivo Langendorff Model
4.5. Molecular Docking of cTnI in δPKC
4.6. Computational Analyses
4.7. Western Blot Analysis
4.8. Mitochondrial Function
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Ioacara, S.; Popescu, A.C.; Tenenbaum, J.; Dimulescu, D.R.; Popescu, M.R.; Sirbu, A.; Fica, S. Acute Myocardial Infarction Mortality Rates and Trends in Romania between 1994 and 2017. Int. J. Environ. Res. Public Health 2019, 17, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Alam, P.; Maliken, B.D.; Jones, S.M.; Ivey, M.J.; Wu, Z.; Wang, Y.; Kanisicak, O. Cardiac Remodeling and Repair: Recent Approaches, Advancements, and Future Perspective. Int. J. Mol. Sci. 2021, 22, 13104. [Google Scholar] [CrossRef]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic opportunities for senolysis in cardiovascular disease. FEBS J. 2022. [Google Scholar] [CrossRef]
- Adameova, A.; Horvath, C.; Abdul-Ghani, S.; Varga, Z.V.; Suleiman, M.S.; Dhalla, N.S. Interplay of Oxidative Stress and Necrosis-like Cell Death in Cardiac Ischemia/Reperfusion Injury: A Focus on Necroptosis. Biomedicines 2022, 10, 127. [Google Scholar] [CrossRef]
- Bright, R.; Raval, A.P.; Dembner, J.M.; Perez-Pinzon, M.A.; Steinberg, G.K.; Yenari, M.A.; Mochly-Rosen, D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 6880–6888. [Google Scholar] [CrossRef]
- Churchill, E.N.; Murriel, C.L.; Chen, C.H.; Mochly-Rosen, D.; Szweda, L.I. Reperfusion-induced translocation of deltaPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ. Res. 2005, 97, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Qvit, N.; Kornfeld, O.S.; Mochly-Rosen, D. Engineered Substrate-Specific Delta PKC Antagonists to Enhance Cardiac Therapeutics. Angew. Chem. 2016, 55, 15672–15679. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.W.; Hinken, A.C.; Patrick, S.E.; Solaro, R.J.; Kobayashi, T. Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J. Biol. Chem. 2010, 285, 11810–11817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkart, E.M.; Sumandea, M.P.; Kobayashi, T.; Nili, M.; Martin, A.F.; Homsher, E.; Solaro, R.J. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 2003, 278, 11265–11272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souroujon, M.C.; Mochly-Rosen, D. Peptide modulators of protein-protein interactions in intracellular signaling. Nat. Biotechnol. 1998, 16, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Takeda, S.; Oda, T.; Fujiwara, S. Structures of the troponin core domain containing the cardiomyopathy-causing mutants studied by small-angle X-ray scattering. Biophys. Phys. 2015, 12, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. 1997, 273, H1544–H1554. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nino, W.R.; Zazueta, C.; Buelna-Chontal, M.; Silva-Palacios, A. Mitochondrial Quality Control in Cardiac-Conditioning Strategies against Ischemia-Reperfusion Injury. Life 2021, 11, 1123. [Google Scholar] [CrossRef]
- Xin, Y.; Zhang, X.; Li, J.; Gao, H.; Li, J.; Li, J.; Hu, W.; Li, H. New Insights Into the Role of Mitochondria Quality Control in Ischemic Heart Disease. Front. Cardiovasc. Med. 2021, 8, 774619. [Google Scholar] [CrossRef]
- Turkieh, A.; El Masri, Y.; Pinet, F.; Dubois-Deruy, E. Mitophagy Regulation Following Myocardial Infarction. Cells 2022, 11, 199. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Love, M.; Kirshenbaum, L.A. Intersection of autophagy regulation and circadian rhythms in the heart. Biochim. Biophys. Acta. Mol. Basis Dis. 2022, 1868, 166354. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, Y.; Luo, X.; Wang, J.; Lan, X.; Liu, P.; Feng, Y.; Jian, W. Myocardial Ischemia-Reperfusion Injury: Therapeutics from a Mitochondria-Centric Perspective. Cardiology 2021, 146, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Takegawa, R.; Shoaib, M.; Aoki, T.; Choudhary, R.C.; Kuschner, C.E.; Nishikimi, M.; Miyara, S.J.; Rolston, D.M.; Guevara, S.; et al. Mitochondrial transplantation therapy for ischemia reperfusion injury: A systematic review of animal and human studies. J. Transl. Med. 2021, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8, 316. [Google Scholar] [CrossRef] [Green Version]
- Al-Salam, S.; Hashmi, S. Myocardial Ischemia Reperfusion Injury: Apoptotic, Inflammatory and Oxidative Stress Role of Galectin-3. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 50, 1123–1139. [Google Scholar] [CrossRef]
- Haushalter, K.J.; Schilling, J.M.; Song, Y.; Sastri, M.; Perkins, G.A.; Strack, S.; Taylor, S.S.; Patel, H.H. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIalpha. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1231–H1242. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.N.; Vrellaku, B.; Monterisi, S.; Chu, S.M.; Rawlings, N.; Lomas, O.; Marchal, G.A.; Waithe, D.; Syeda, F.; Gajendragadkar, P.R.; et al. Oxidation of Protein Kinase A Regulatory Subunit PKARIalpha Protects Against Myocardial Ischemia-Reperfusion Injury by Inhibiting Lysosomal-Triggered Calcium Release. Circulation 2021, 143, 449–465. [Google Scholar] [CrossRef]
- Zhao, J.; Conklin, D.J.; Guo, Y.; Zhang, X.; Obal, D.; Guo, L.; Jagatheesan, G.; Katragadda, K.; He, L.; Yin, X.; et al. Cardiospecific Overexpression of ATPGD1 (Carnosine Synthase) Increases Histidine Dipeptide Levels and Prevents Myocardial Ischemia Reperfusion Injury. J. Am. Heart Assoc. 2020, 9, e015222. [Google Scholar] [CrossRef]
- Huang, J.; Li, R.; Wang, C. The Role of Mitochondrial Quality Control in Cardiac Ischemia/Reperfusion Injury. Oxid. Med. Cell. Longev. 2021, 2021, 5543452. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamamoto, M.; Nakamura, J.; Mii, A.; Yamamoto, S.; Takahashi, M.; Kaneko, K.; Uchino, E.; Sato, Y.; Fukuma, S.; et al. Spatiotemporal ATP Dynamics during AKI Predict Renal Prognosis. J. Am. Soc. Nephrol. JASN 2020, 31, 2855–2869. [Google Scholar] [CrossRef]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, D.; Guo, M. The roles of PKC-delta and PKC-epsilon in myocardial ischemia/reperfusion injury. Pharmacol. Res. 2021, 170, 105716. [Google Scholar] [CrossRef] [PubMed]
- Pyle, W.G.; Sumandea, M.P.; Solaro, R.J.; De Tombe, P.P. Troponin I serines 43/45 and regulation of cardiac myofilament function. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1215–H1224. [Google Scholar] [CrossRef] [Green Version]
- Pi, Y.; Kemnitz, K.R.; Zhang, D.; Kranias, E.G.; Walker, J.W. Phosphorylation of troponin I controls cardiac twitch dynamics: Evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ. Res. 2002, 90, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, D.E.; Wolska, B.M.; Pyle, W.G.; Roman, B.B.; Dowell, J.C.; Buttrick, P.M.; Koretsky, A.P.; Del Nido, P.; Solaro, R.J. alpha-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2397–H2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, B.B.; Goldspink, P.H.; Spaite, E.; Urboniene, D.; McKinney, R.; Geenen, D.L.; Solaro, R.J.; Buttrick, P.M. Inhibition of PKC phosphorylation of cTnI improves cardiac performance in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2089–H2095. [Google Scholar] [CrossRef]
- Chen, C.L.; Zhang, L.; Jin, Z.; Kasumov, T.; Chen, Y.R. Mitochondrial redox regulation and myocardial ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2022, 322, C12–C23. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Choi, J.; Yun, J.S.; Song, H.; Kim, N.H.; Kim, H.S.; Yook, J.I. Exploring the chemical space of protein-protein interaction inhibitors through machine learning. Sci. Rep. 2021, 11, 13369. [Google Scholar] [CrossRef]
- Cheng, S.S.; Yang, G.J.; Wang, W.; Leung, C.H.; Ma, D.L. The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 2020, 13, 26. [Google Scholar] [CrossRef]
- Sawyer, A. Developing drugs for the ‘undruggable’. BioTechniques 2020, 69, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Schueler-Furman, O. Druggable protein-protein interactions--from hot spots to hot segments. Curr. Opin. Chem. Biol. 2013, 17, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Neduva, V.; Russell, R.B. Peptides mediating interaction networks: New leads at last. Curr. Opin. Biotechnol. 2006, 17, 465–471. [Google Scholar] [CrossRef]
- Meyer, K.; Selbach, M. Peptide-based Interaction Proteomics. Mol. Cell. Proteom. MCP 2020, 19, 1070–1075. [Google Scholar] [CrossRef]
- Kannan, S.; Aronica, P.G.A.; Ng, S.; Gek Lian, D.T.; Frosi, Y.; Chee, S.; Shimin, J.; Yuen, T.Y.; Sadruddin, A.; Kaan, H.Y.K.; et al. Macrocyclization of an all-d linear alpha-helical peptide imparts cellular permeability. Chem. Sci. 2020, 11, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc. Natl. Acad. Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qvit, N.; Lin, A.J.; Elezaby, A.; Ostberg, N.P.; Campos, J.C.; Ferreira, J.C.B.; Mochly-Rosen, D. A Selective Inhibitor of Cardiac Troponin I Phosphorylation by Delta Protein Kinase C (δPKC) as a Treatment for Ischemia-Reperfusion Injury. Pharmaceuticals 2022, 15, 271. https://doi.org/10.3390/ph15030271
Qvit N, Lin AJ, Elezaby A, Ostberg NP, Campos JC, Ferreira JCB, Mochly-Rosen D. A Selective Inhibitor of Cardiac Troponin I Phosphorylation by Delta Protein Kinase C (δPKC) as a Treatment for Ischemia-Reperfusion Injury. Pharmaceuticals. 2022; 15(3):271. https://doi.org/10.3390/ph15030271
Chicago/Turabian StyleQvit, Nir, Amanda J. Lin, Aly Elezaby, Nicolai P. Ostberg, Juliane C. Campos, Julio C. B. Ferreira, and Daria Mochly-Rosen. 2022. "A Selective Inhibitor of Cardiac Troponin I Phosphorylation by Delta Protein Kinase C (δPKC) as a Treatment for Ischemia-Reperfusion Injury" Pharmaceuticals 15, no. 3: 271. https://doi.org/10.3390/ph15030271