
AAZTA-Derived Chelators for the Design of Innovative Radiopharmaceuticals with Theranostic Applications

 , , and
, , and
Abstract
:1. Introduction
2. Design, Synthesis, and Kinetic Properties
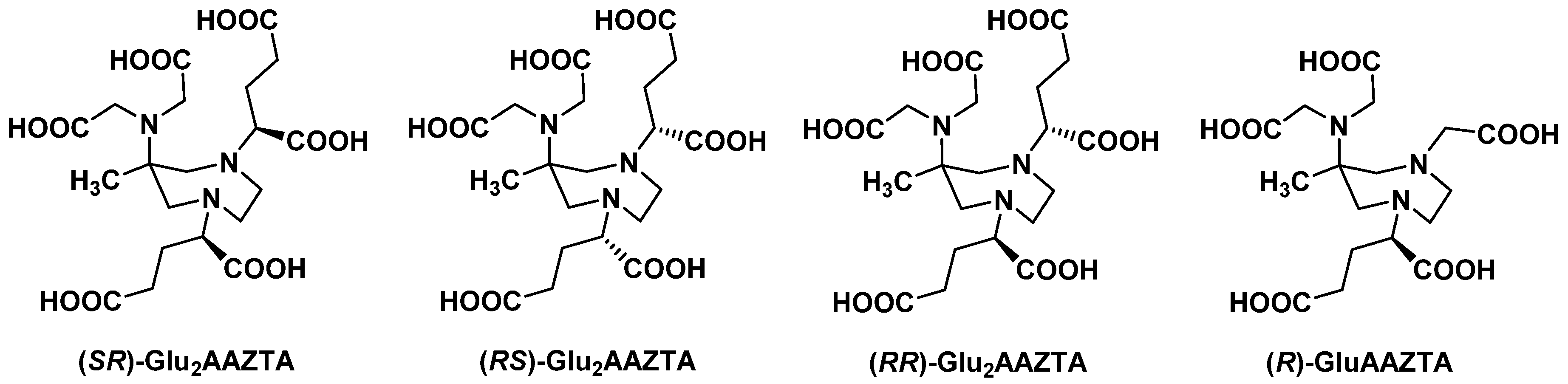
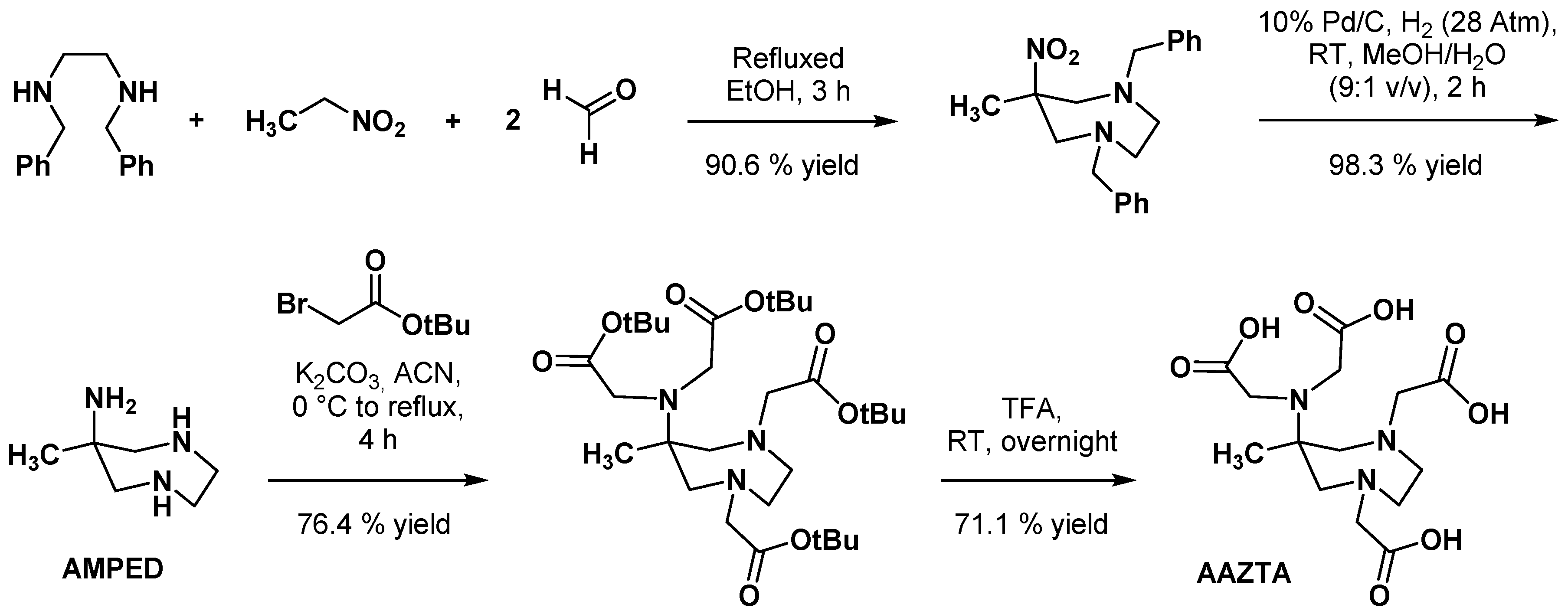
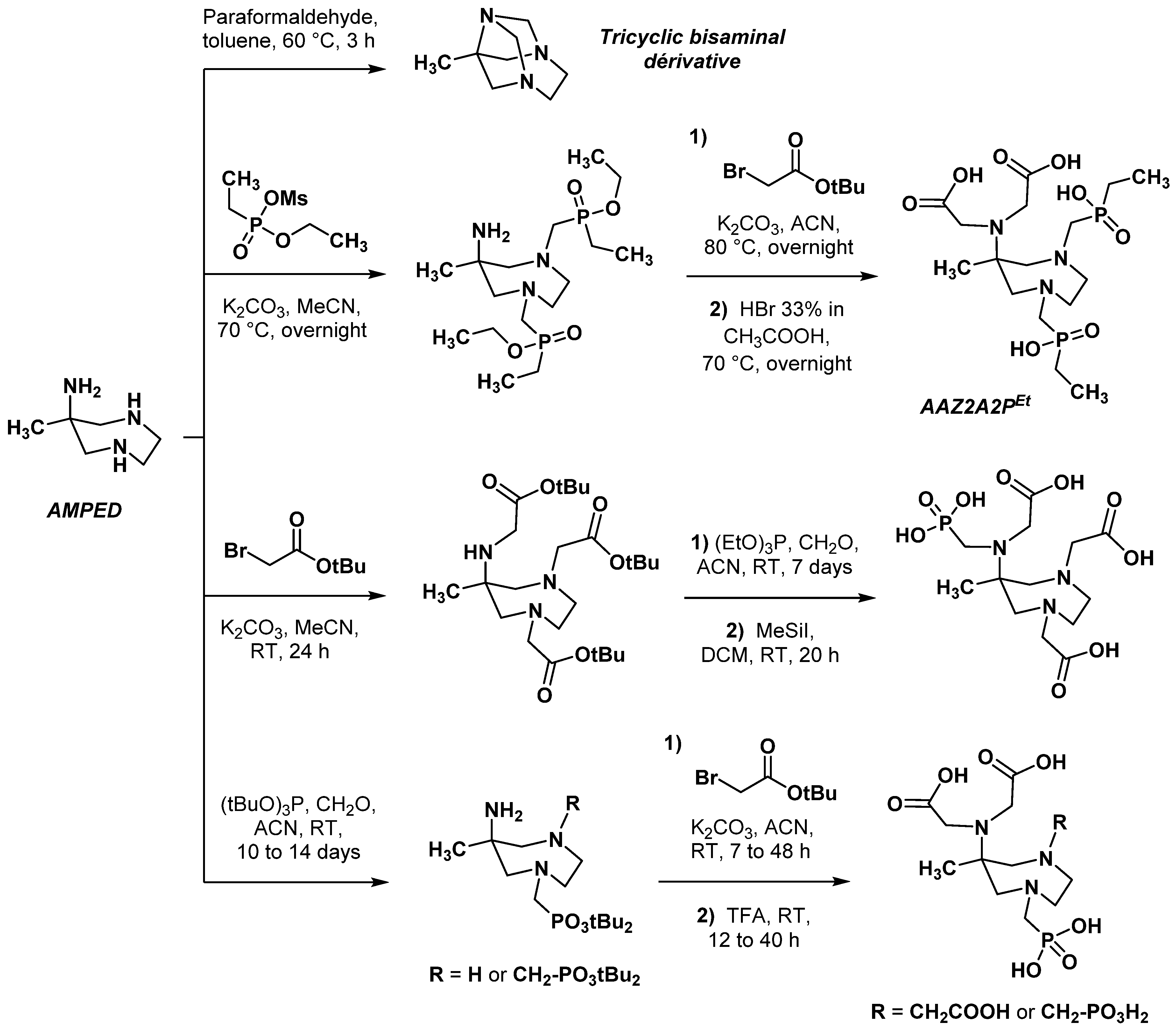
2.1. Original AAZTA Derivatives
2.2. AAZTA Modulations to Improve Coordination Behavior
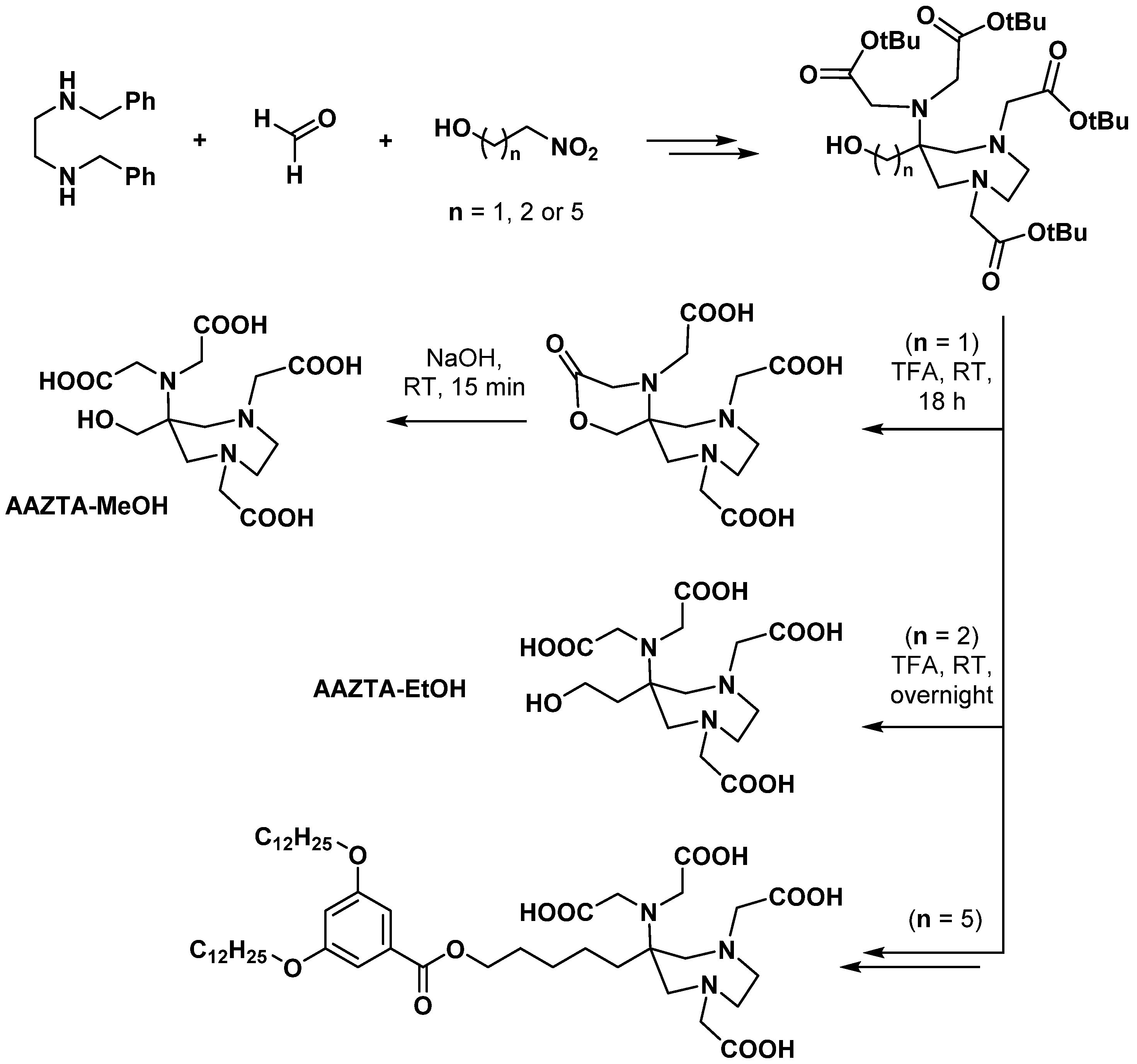
2.2.1. Modifications in the Number and Nature of Coordinating Side Arms
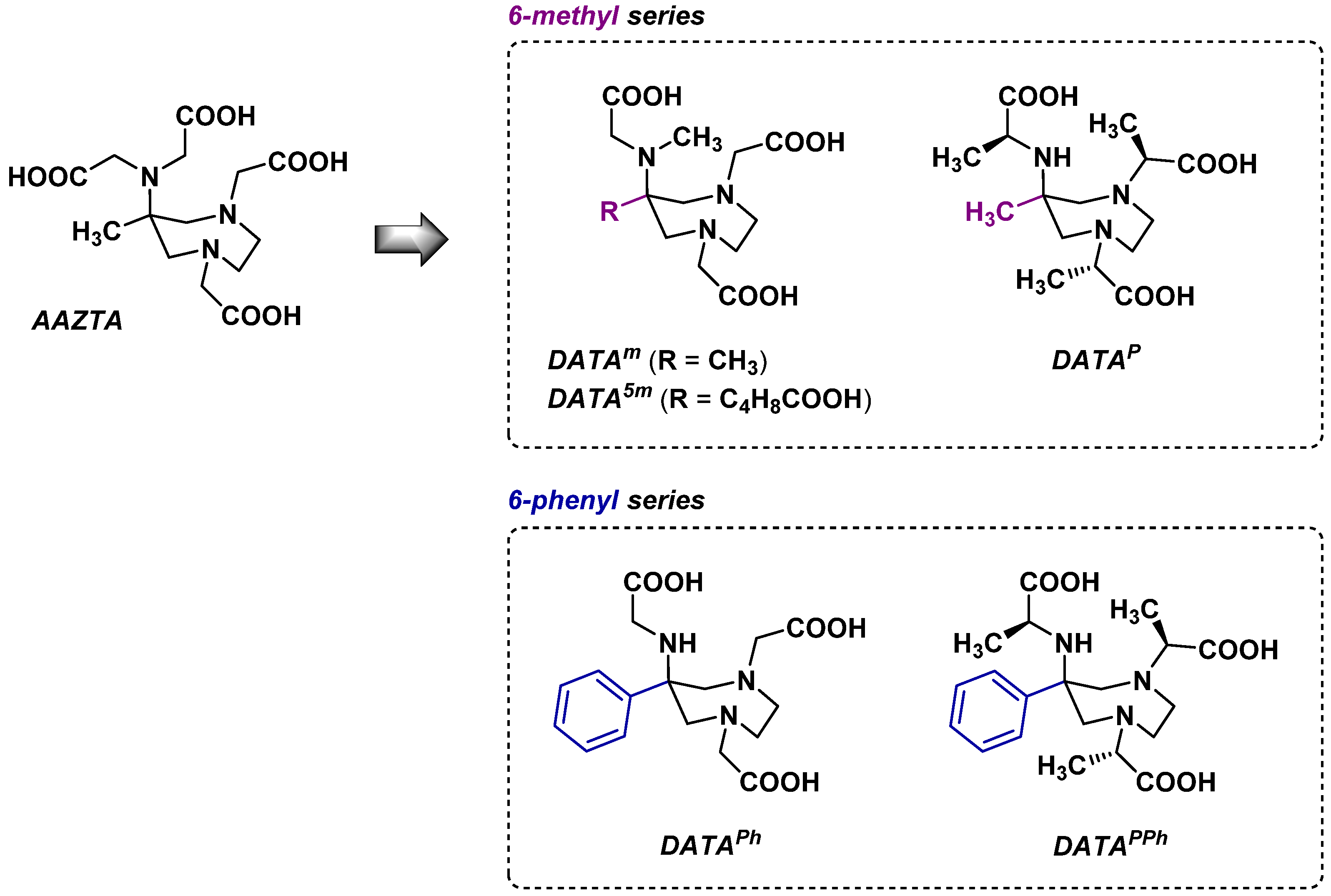
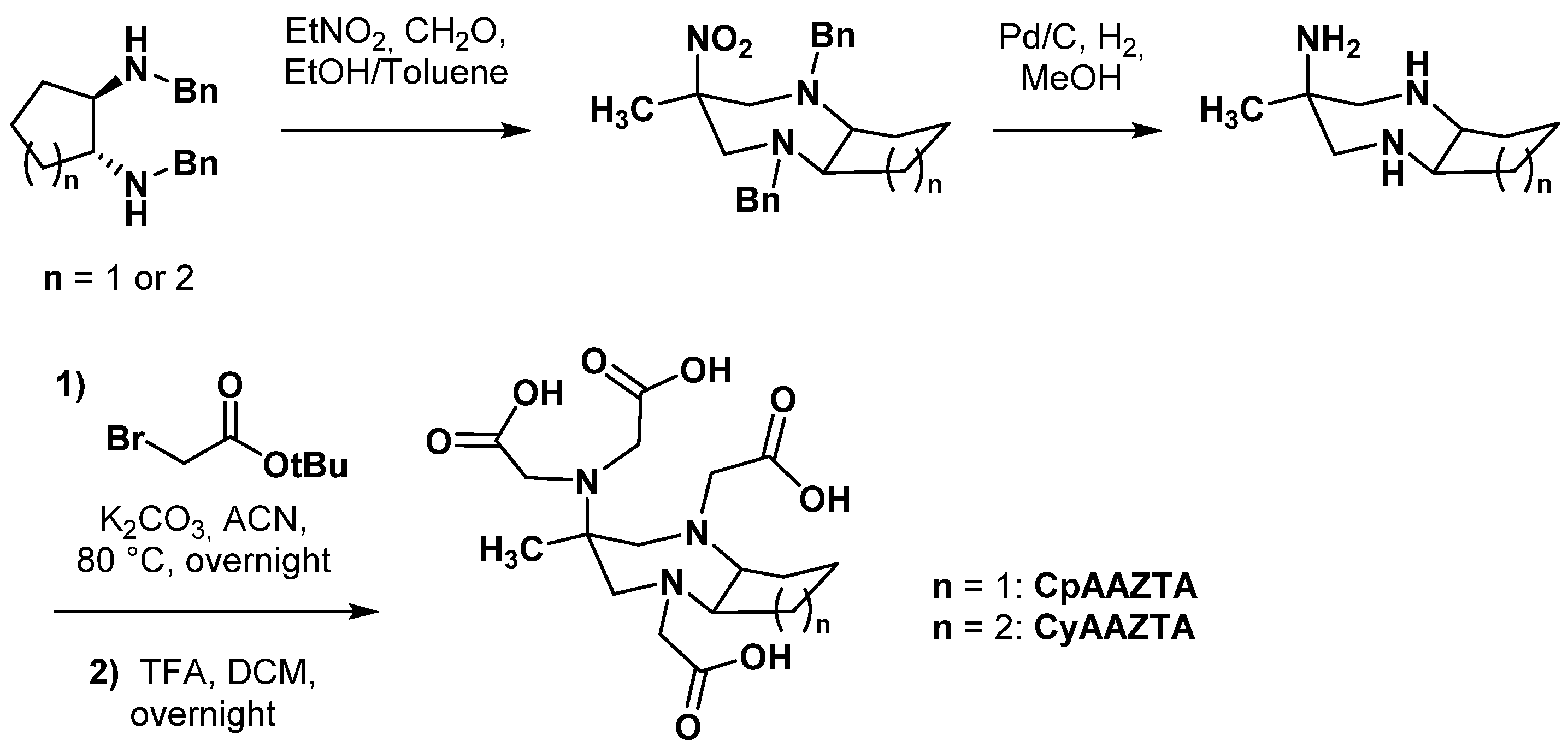
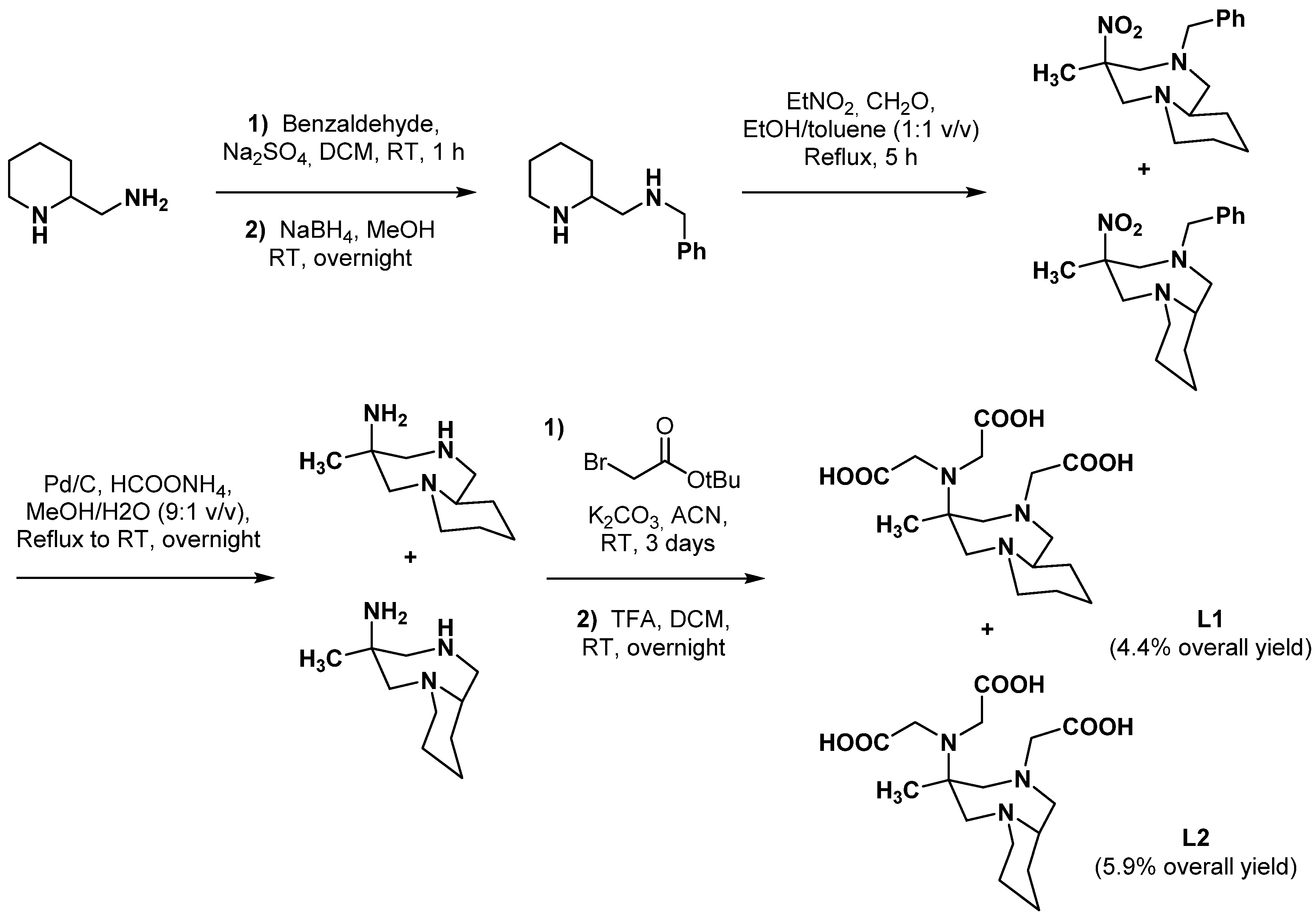
2.2.2. Backbone Rigidification
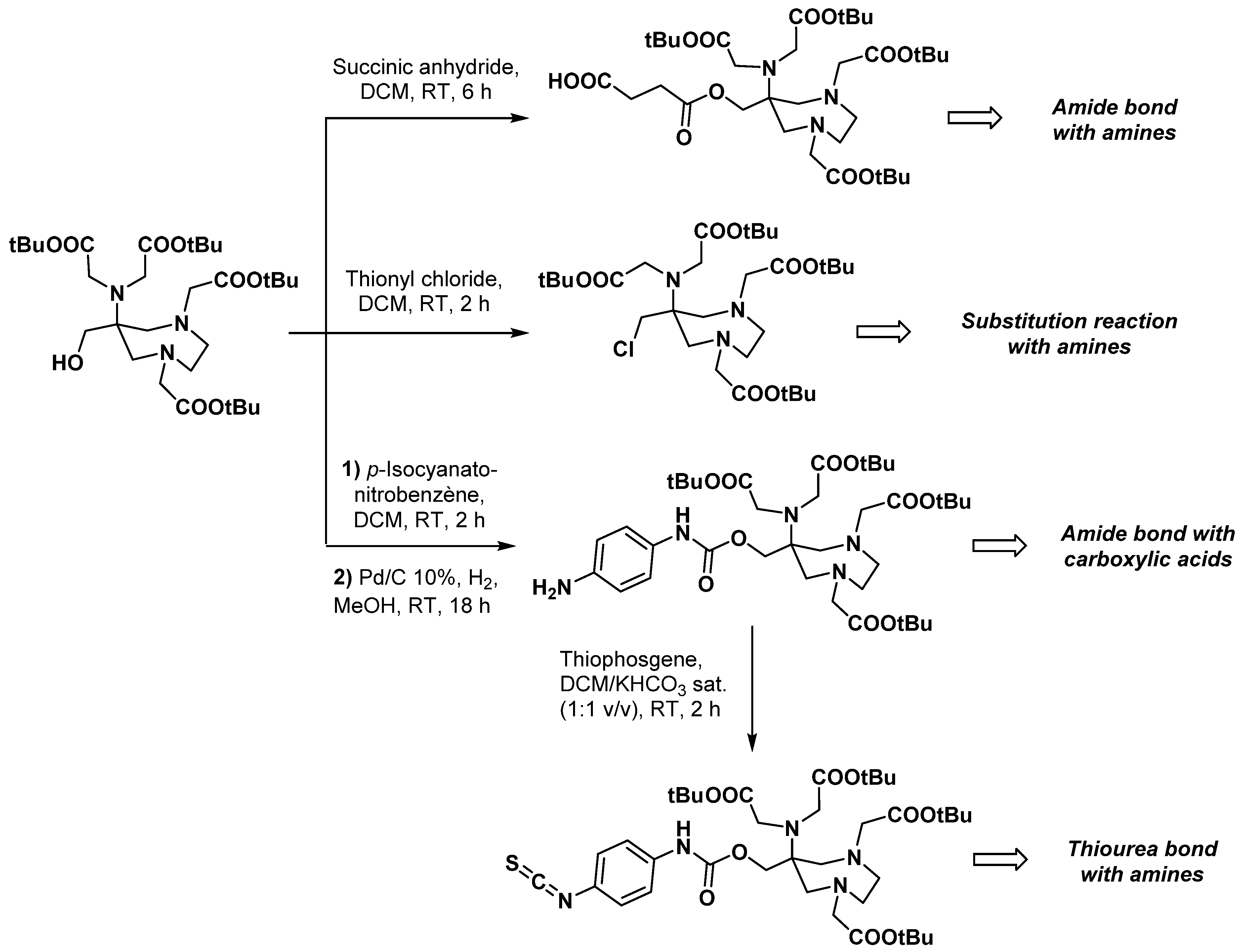
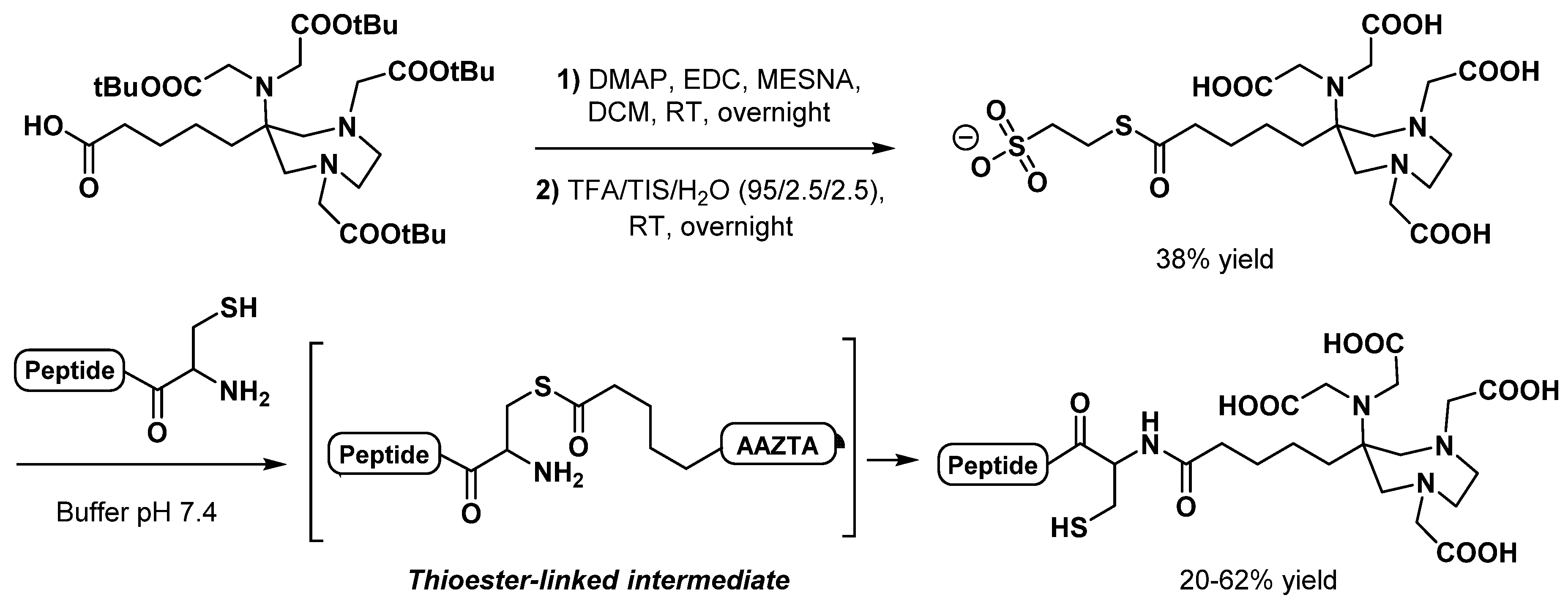
2.3. AAZTA Modulations to Obtain Bifunctional Chelating Agents (BCA)
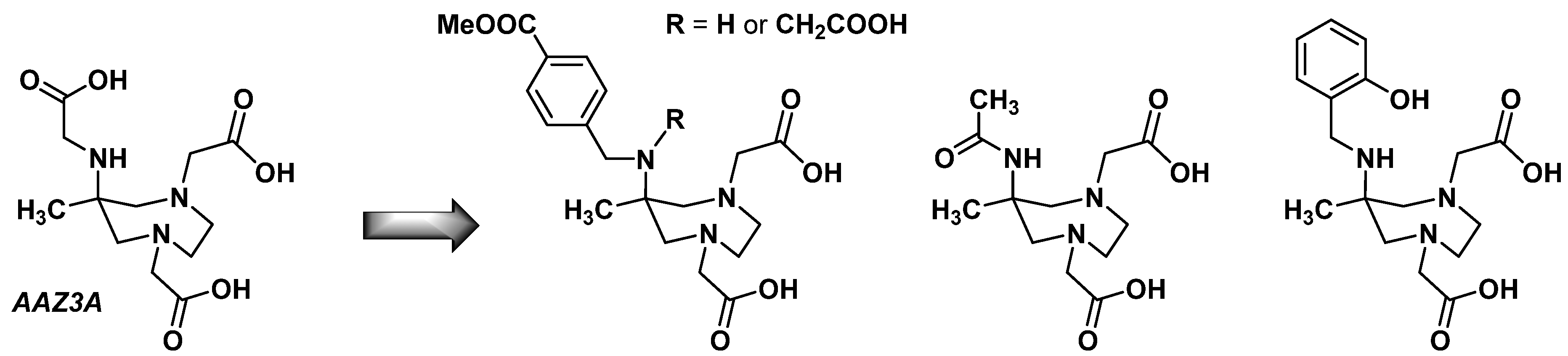
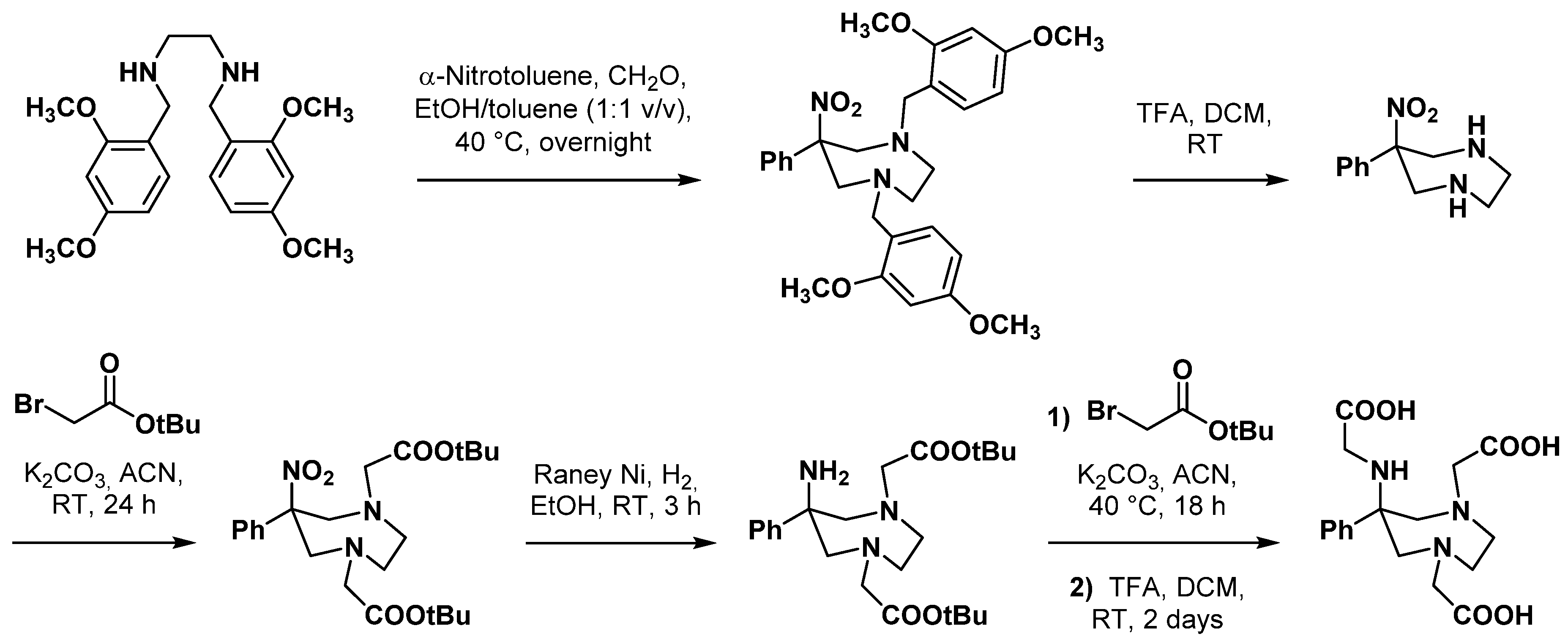
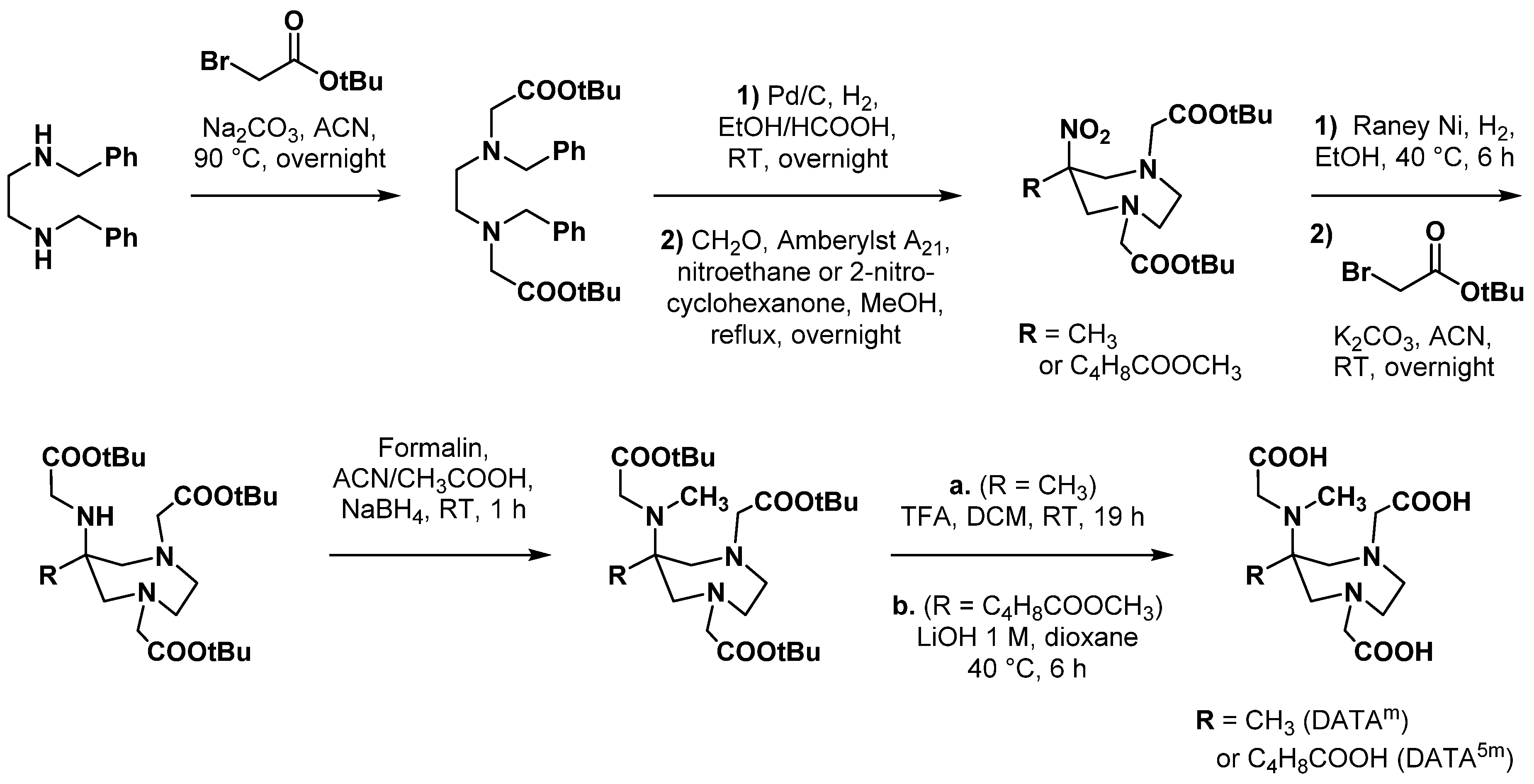
2.4. AAZ3A Derivatives
3. Applications in Nuclear Medicine
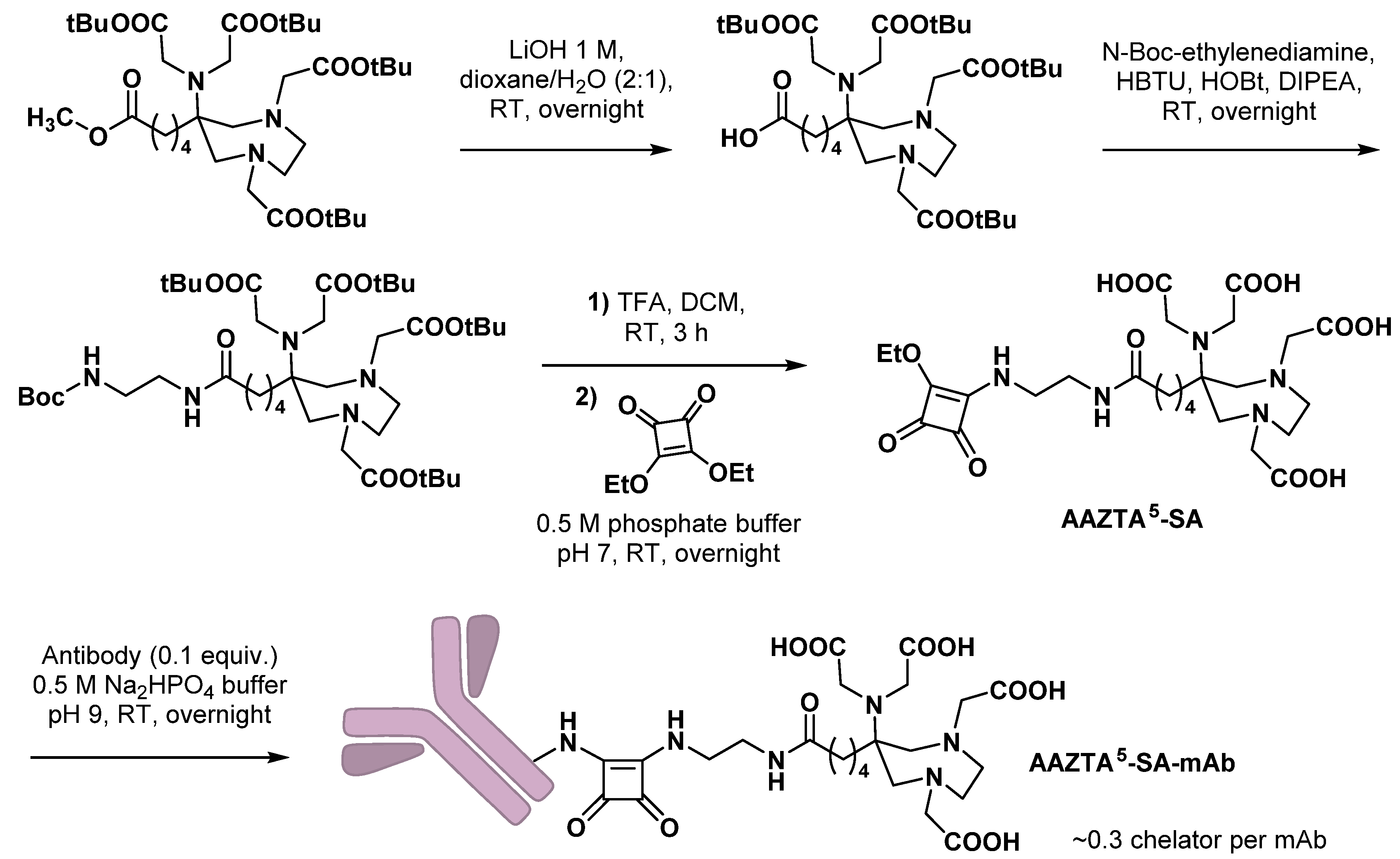
3.1. Monoclonal Antibodies
3.2. Small Molecules
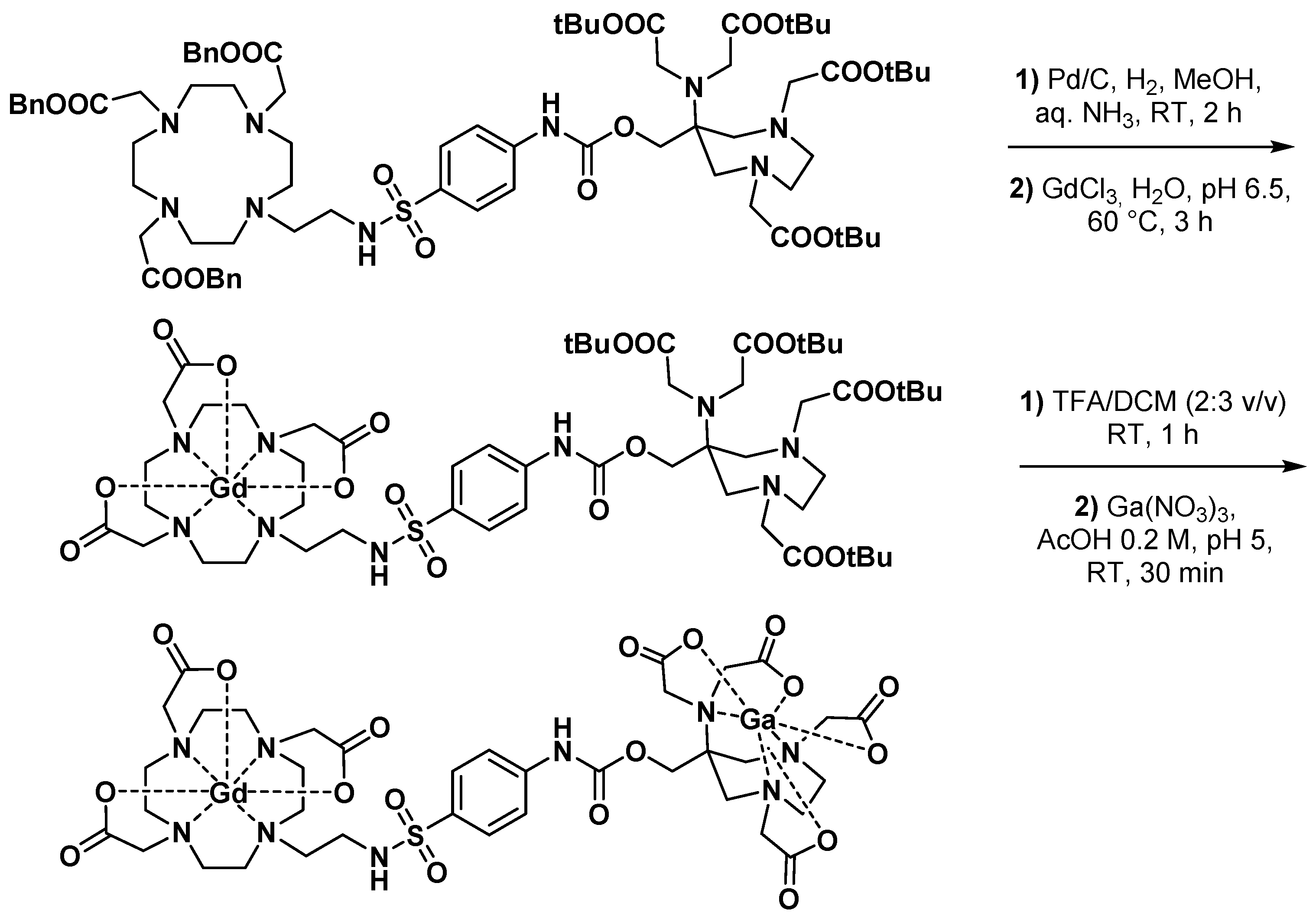
3.2.1. Dual MRI/PET Imaging Agents
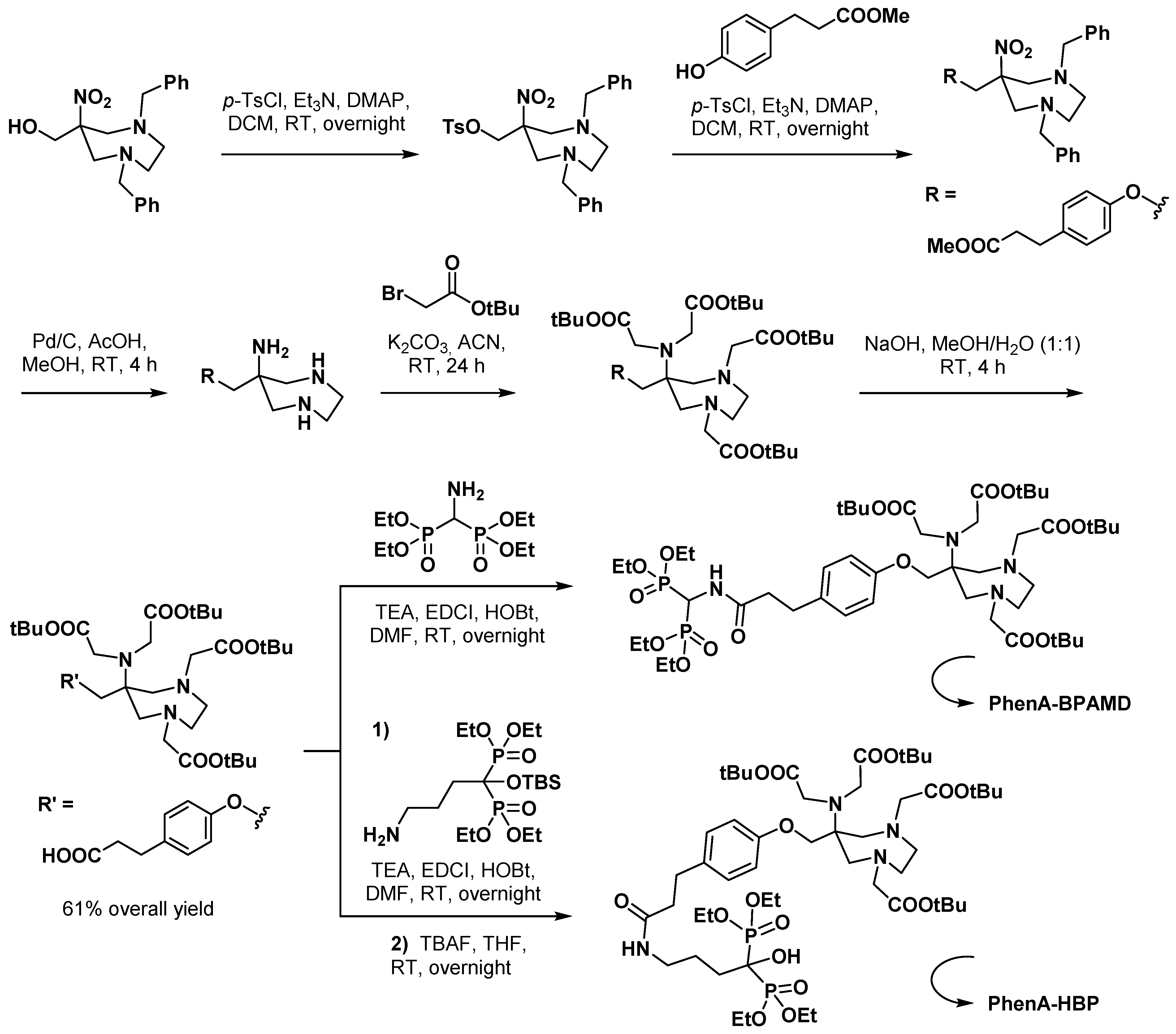
3.2.2. Bisphosphonate Derivatives
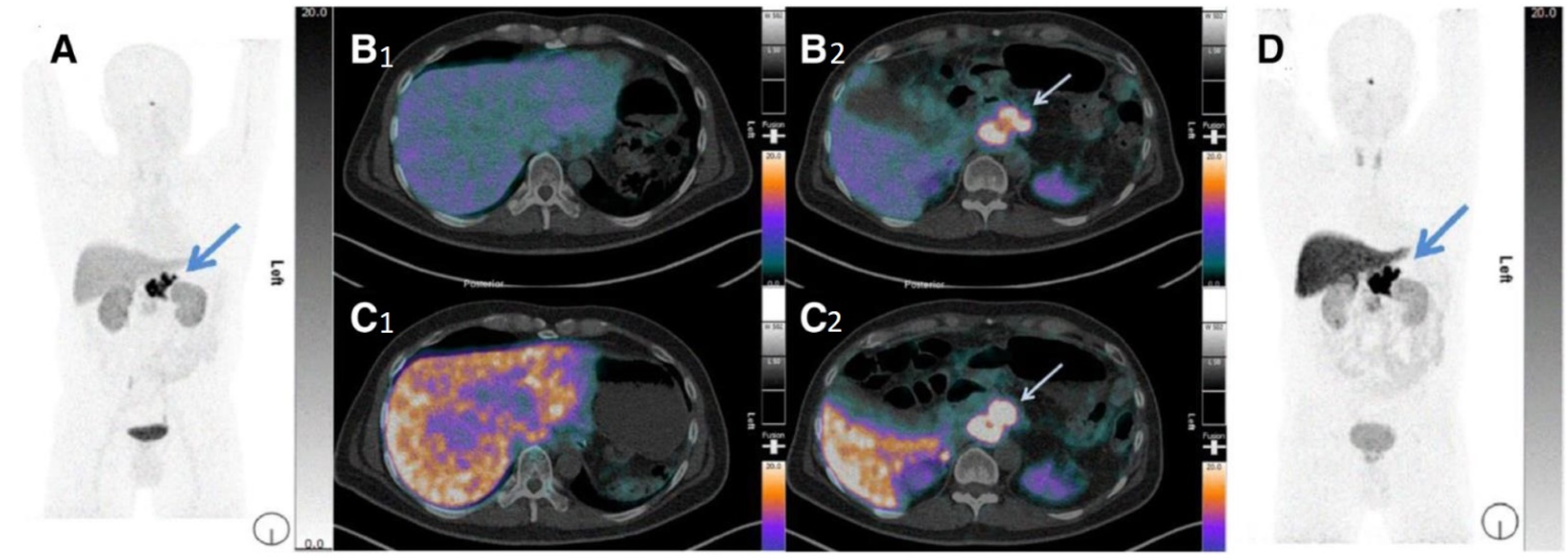
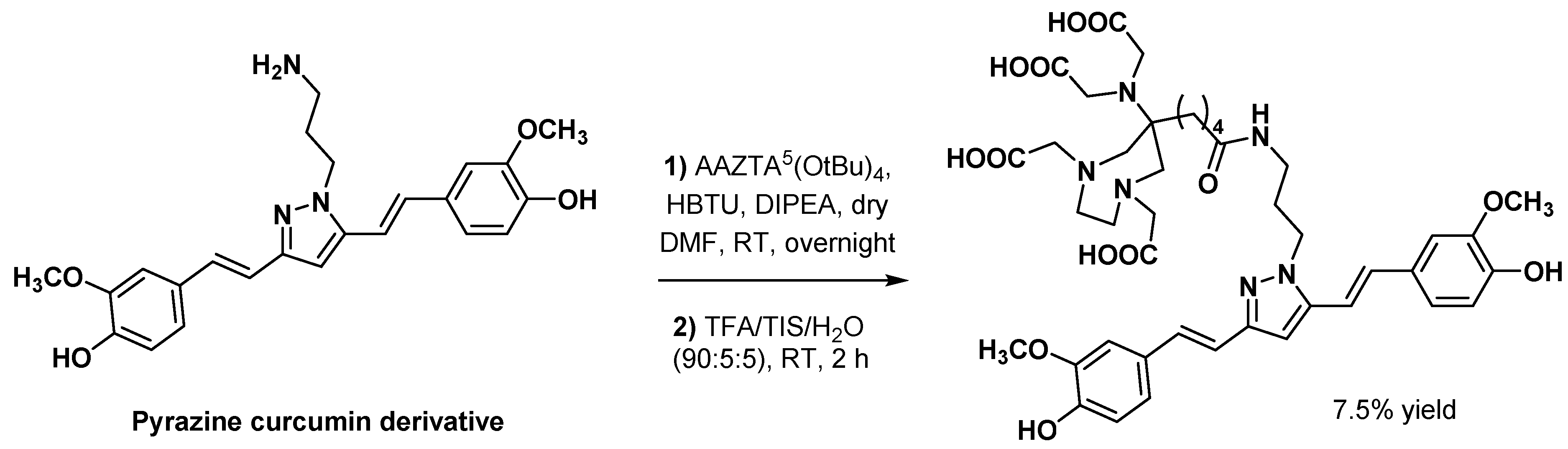
3.2.3. Curcumin Derivatives
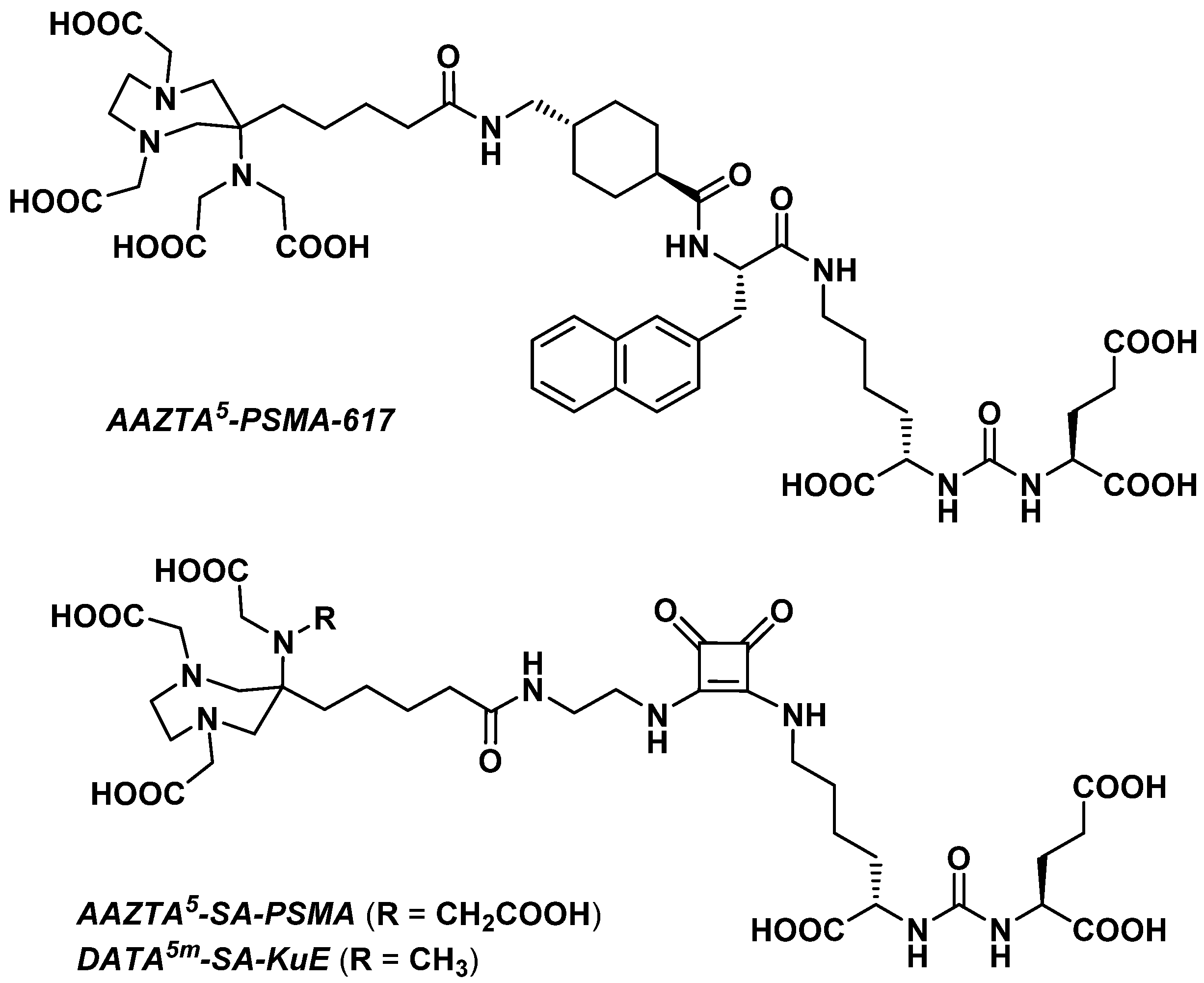
3.2.4. Prostate-Specific Antigen Ligands
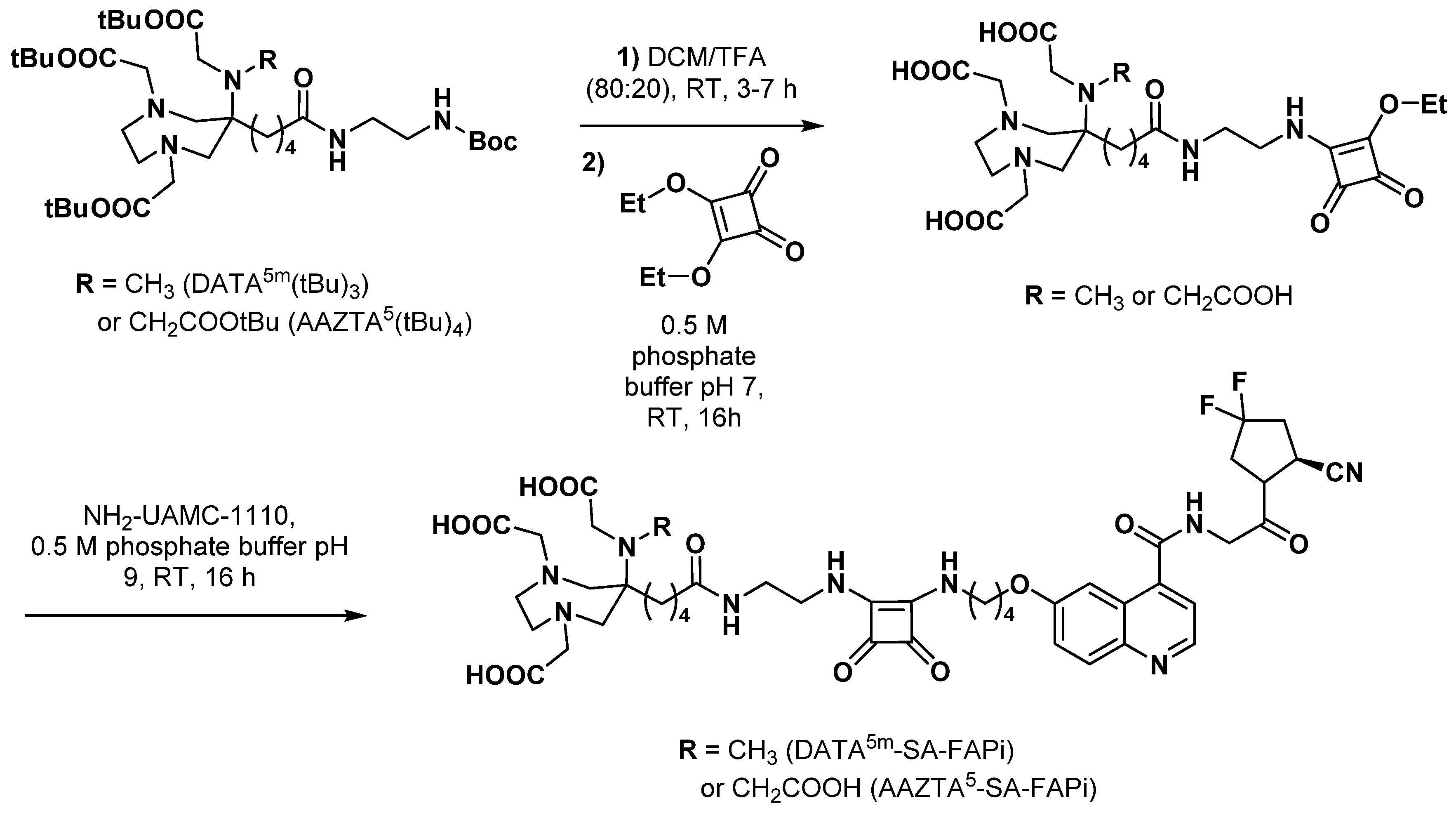
3.2.5. Fibroblast Activation Protein Inhibitors
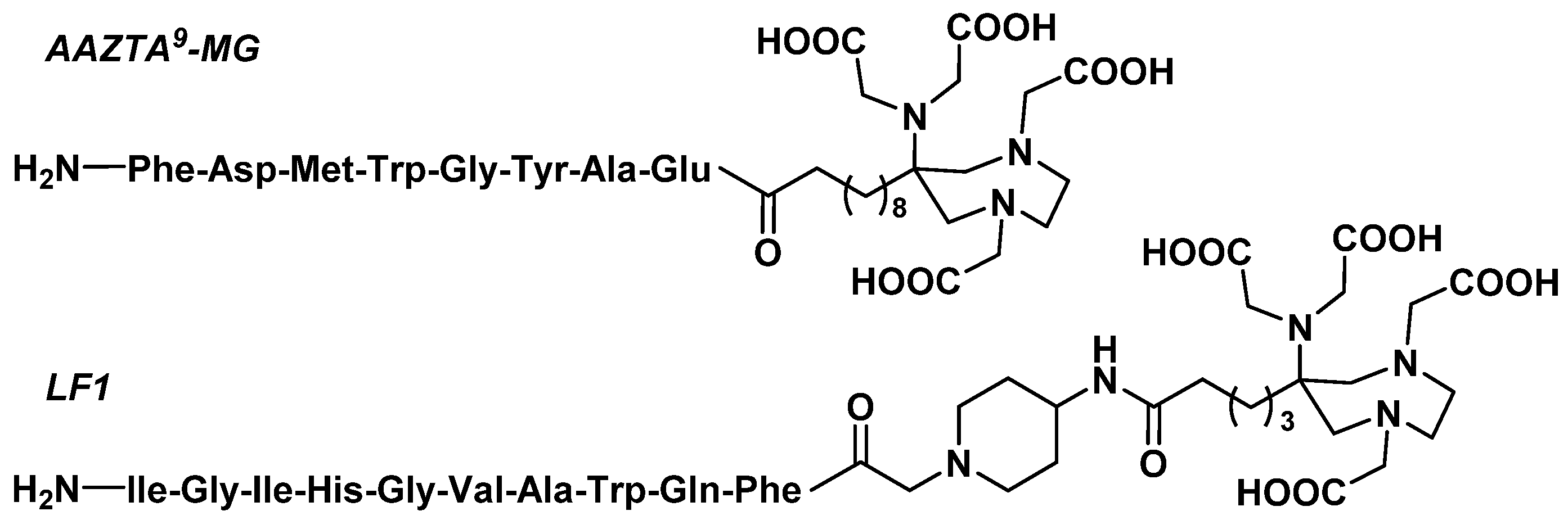
3.3. Peptides
3.3.1. RGD Peptides
3.3.2. Gastrin Analogues
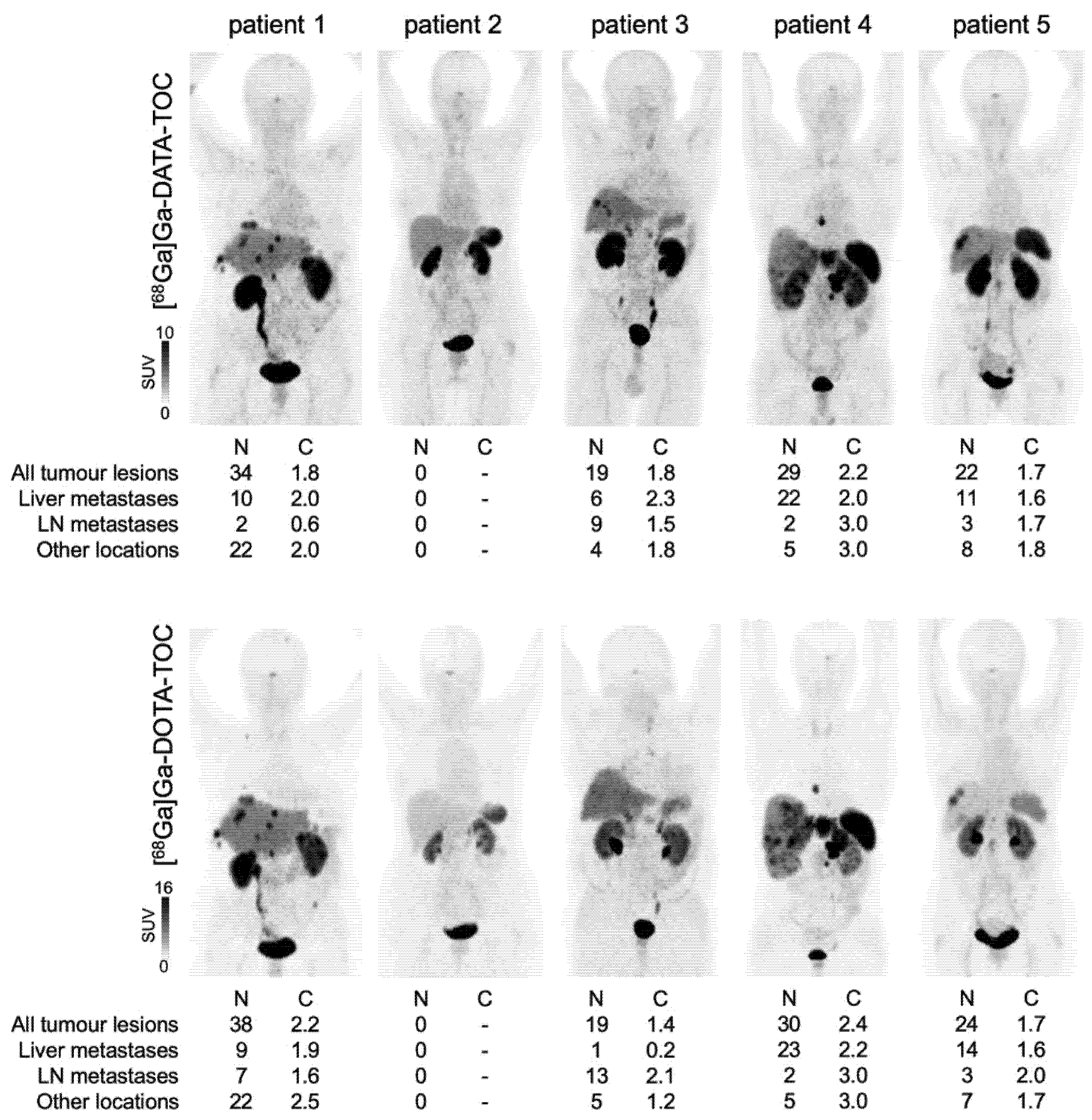
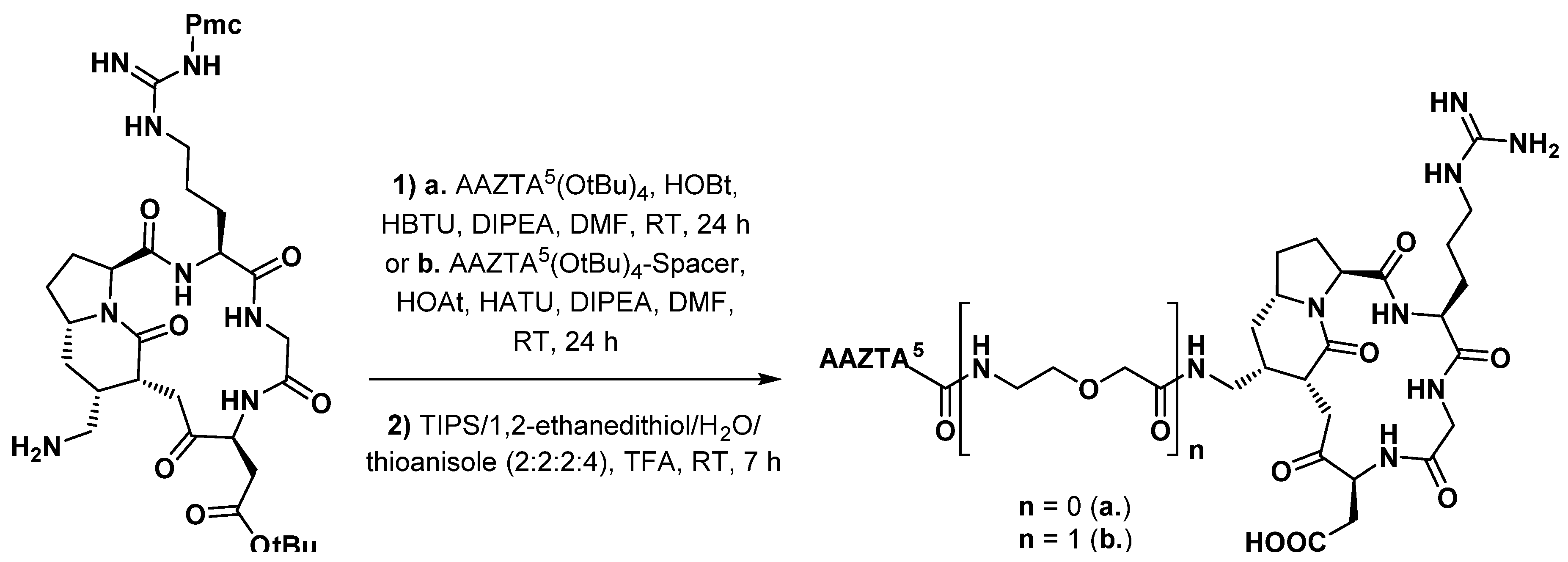
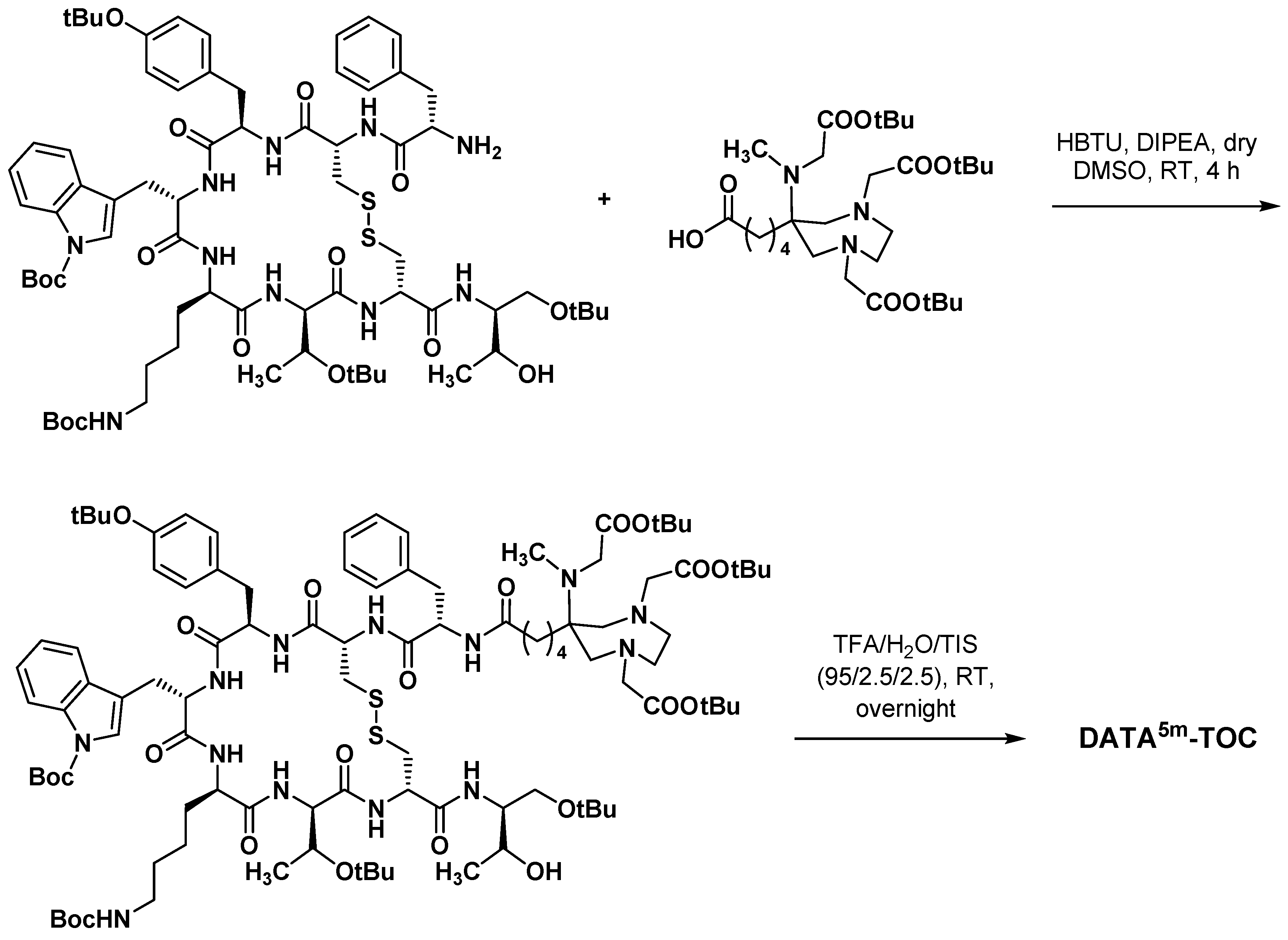
3.3.3. Octreotide Analogues
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelkar, S.S.; Reineke, T.M. Theranostics: Combining imaging and therapy. Bioconjug. Chem. 2011, 22, 1879–1903. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chen, X.; Si, J.; Mou, X.; Dong, X. All-in-one nanomedicine: Multifunctional single-component nanoparticles for cancer theranostics. Small 2021, 17, 2103072. [Google Scholar] [CrossRef] [PubMed]
- Kharbikar, B.N.; Zhong, J.X.; Cuylear, D.L.; Perez, C.A.; Desai, T.A. Theranostic biomaterials for tissue engineering. Curr. Opin. Biomed. Eng. 2021, 19, 100299. [Google Scholar] [CrossRef]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of theranostics: An outlook on precision oncology in Nuclear Medicine. J. Nucl. Med. 2019, 60, 13S–19S. [Google Scholar] [CrossRef] [Green Version]
- Hertz, S. Radioactive iodine in the study of thyroid physiology: VII. The use of radioactive iodine therapy in hyperthyroidism. JAMA 1946, 131, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Seidlin, S.M. Radioactive iodine therapy: Effect on functioning metastases of adenocarcinoma of the thyroid. JAMA 1946, 132, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Khreish, F.; Ghazal, Z.; Marlowe, R.J.; Rosar, F.; Sabet, A.; Maus, S.; Linxweiler, J.; Bartholomä, M.; Ezziddin, S. 177Lu-PSMA-617 radioligand therapy of metastatic castration-resistant prostate cancer: Initial 254-patient results from a prospective registry (REALITY Study). Eur. J. Nucl. Med. Mol. Imaging 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Mikolajczak, R.; Huclier-Markai, S.; Alliot, C.; Haddad, F.; Szikra, D.; Forgacs, V.; Garnuszek, P. Production of scandium radionuclides for theranostic applications: Towards standardization of quality requirements. EJNMMI Radiopharm. Chem. 2021, 6, 19. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Aime, S.; Calabi, L.; Cavallotti, C.; Gianolio, E.; Giovenzana, G.B.; Losi, P.; Maiocchi, A.; Palmisano, G.; Sisti, M. [Gd-AAZTA]: A new structural entry for an improved generation of MRI contrast agents. Inorg. Chem. 2004, 43, 7588–7590. [Google Scholar] [CrossRef]
- Baranyai, Z.; Uggeri, F.; Giovenzana, G.B.; Bényei, A.; Brücher, E.; Aime, S. Equilibrium and kinetic properties of the lanthanoids(III) and various divalent metal complexes of the heptadentate ligand AAZTA. Chem. Eur. J. 2009, 15, 1696–1705. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Tircsó, G. Kinetic inertness of the Mn2+ complexes formed with AAZTA and some open-chain EDTA derivatives. Inorg. Chem. 2012, 51, 10065–10067. [Google Scholar] [CrossRef] [Green Version]
- Baranyai, Z.; Uggeri, F.; Maiocchi, A.; Giovenzana, G.B.; Cavallotti, C.; Takács, A.; Tóth, I.; Bányai, I.; Bényei, A.; Brucher, E.; et al. Equilibrium, kinetic and structural studies of AAZTA complexes with Ga3+, In3+ and Cu2+. Eur. J. Inorg. Chem. 2013, 2013, 147–162. [Google Scholar] [CrossRef]
- Huclier-Markai, S.; Alliot, C.; Kerdjoudj, R.; Mougin-Degraef, M.; Chouin, N.; Haddad, F. Promising scandium radionuclides for Nuclear Medicine: A review on the production and chemistry up to in vivo proofs of concept. Cancer Biother. Radiopharm. 2018, 33, 316–329. [Google Scholar] [CrossRef]
- Nagy, G.; Szikra, D.; Trencsényi, G.; Fekete, A.; Garai, I.; Giani, A.M.; Negri, R.; Masciocchi, N.; Maiocchi, A.; Uggeri, F.; et al. AAZTA: An ideal chelating agent for the development of 44Sc PET imaging agents. Angew. Chem. 2017, 56, 2118–2122. [Google Scholar] [CrossRef]
- Mathias, C.J.; Sun, Y.; Welch, M.J.; Connett, J.M.; Philpott, G.W.; Martell, A.E. N,N′-bis(2-hydroxybenzyl)-1-(4-bromoacetamidobenzyl)-1,2-ethylene-diamine-N,N′-diacetic acid: A new bifunctional chelate for radio-labeling antibodies. Bioconjug. Chem. 1990, 1, 204–211. [Google Scholar] [CrossRef]
- Eder, M.; Wängler, B.; Knackmuss, S.; LeGall, F.; Little, M.; Haberkorn, M.; Mier, W.; Eisenhut, M. Tetrafluorophenolate of HBED-CC: A versatile conjugation agent for 68Ga-labeled small recombinant antibodies. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1878–1886. [Google Scholar] [CrossRef]
- Hennrich, U.; Eder, M. [68Ga]Ga-PSMA-11: The first FDA-approved 68Ga-radiopharmaceutical for PET imaging of prostate cancer. Pharmaceuticals 2021, 14, 713. [Google Scholar] [CrossRef]
- Ermelindo, A.; Gambino, G.; Tei, L. Synthesis of a mixed carboxylate–phosphinate AAZTA-like ligand and relaxometric characterization of its GdIII complex. Tetrahedron Lett. 2013, 54, 6378–6380. [Google Scholar] [CrossRef]
- Elemento, E.M.; Parker, D.; Aime, S.; Gianolio, E.; Lattuada, L. Variation of water exchange dynamics with ligand structure and stereochemistry in lanthanide complexes based on 1,4-diazepine derivatives. Org. Biomol. Chem. 2009, 7, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Guanci, C.; Pinalli, R.; Aime, S.; Gianolio, E.; Lattuada, L.; Giovenzana, G.B. Synthesis of phosphonic analogues of AAZTA and relaxometric evaluation of the corresponding Gd(III) complexes as potential MRI contrast agents. Tetrahedron Lett. 2015, 56, 1994–1997. [Google Scholar] [CrossRef]
- Notni, J.; Šimeček, J.; Hermann, P.; Wester, H.-J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chem. Eur. J. 2011, 17, 14718–14722. [Google Scholar] [CrossRef] [PubMed]
- Šimeček, J.; Schulz, M.; Notni, J.; Plutnar, J.; Kubíček, V.; Havlíčková, J.; Hermann, P. Complexation of metal ions with TRAP (1,4,7-triazacyclononane phosphinic acid) ligands and 1,4,7-triazacyclononane-1,4,7-triacetic acid: Phosphinate-containing ligands as unique chelators for trivalent gallium. Inorg. Chem. 2012, 51, 577–590. [Google Scholar] [CrossRef]
- Šimeček, J.; Zemek, O.; Hermann, P.; Wester, H.-J.; Notni, J. A monoreactive bifunctional triazacyclononane phosphinate chelator with high selectivity for gallium-68. ChemMedChem 2012, 7, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Guanci, C.; Giovenzana, G.; Lattuada, L.; Platas-Iglesias, C.; Charbonnière, L.J. AMPED: A new platform for picolinate based luminescent lanthanide chelates. Dalton Trans. 2015, 44, 7654–7661. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Zeglis, B.M.; Cawthray, J.F.; Ramogida, C.F.; Ramos, N.; Lewis, J.S.; Adam, M.J.; Orvig, C. H4Octapa-Trastuzumab: Versatile acyclic chelate system for 111In and 177Lu imaging and therapy. J. Am. Chem. Soc. 2013, 135, 12707–12721. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Domnanich, K.A.; Umbricht, C.A.; van der Meulen, N.P. Scandium and terbium radionuclides for radiotheranostics: Current state of development towards clinical application. Br. J. Radiol. 2018, 91, 20180074. [Google Scholar] [CrossRef]
- Martinelli, J.; Remotti, D.; Tei, L. Selective functionalization of 6-amino-6-methyl-1,4-perhydrodiazepine for the synthesis of a library of polydentate chelators. Org. Biomol. Chem. 2020, 18, 5245–5252. [Google Scholar] [CrossRef]
- Camera, L.; Kinuya, S.; Garmestani, K.; Wu, C.; Brechbiel, M.W.; Pai, L.H.; McMurry, T.J.; Gansow, O.A.; Pastan, I.; Paik, C.H. Evaluation of the serum stability and in vivo biodistribution of CHX-DTPA and other ligands for yttrium labeling of monoclonal antibodies. J. Nucl. Med. 1994, 35, 882–889. [Google Scholar]
- Stimmel, J.B.; Kull, F.C. Samarium-153 and lutetium-177 chelation properties of selected macrocyclic and acyclic ligands. Nucl. Med. Biol. 1998, 25, 117–125. [Google Scholar] [CrossRef]
- Vágner, A.; D’Alessandria, C.; Gambino, G.; Schwaiger, M.; Aime, S.; Maiocchi, A.; Tóth, I.; Baranyai, Z.; Tei, L. A Rigidified AAZTA-like ligand as efficient chelator for 68Ga radiopharmaceuticals. ChemistrySelect 2016, 1, 163–171. [Google Scholar] [CrossRef]
- Vágner, A.; Gianolio, E.; Aime, S.; Maiocchi, A.; Tóth, I.; Baranyai, Z.; Tei, L. High kinetic inertness of a bis-hydrated Gd-complex with a constrained AAZTA-like ligand. Chem. Commun. 2016, 52, 11235–11238. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, J.; Martorana, E.; Tei, L. Synthesis of a rigidified bicyclic AAZTA-like ligand and relaxometric characterization of its GdIII complex. Tetrahedron Lett. 2020, 61, 152573. [Google Scholar] [CrossRef]
- Farkas, E.; Vágner, A.; Negri, R.; Lattuada, L.; Tóth, I.; Colombo, V.; Esteban-Gómez, D.; Platas-Iglesias, C.; Notni, J.; Baranyai, Z.; et al. PIDAZTA: Structurally constrained chelators for the efficient formation of stable gallium-68 complexes at physiological pH. Chem. Eur. J. 2019, 25, 10698–10709. [Google Scholar] [CrossRef]
- Rosa, L.D.; Romanelli, A.; D’Andrea, L.D. Introduction to chemical ligation reactions. In Chemical Ligation; D’Andrea, L.D., Romanelli, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–87. ISBN 978-1-119-04411-6. [Google Scholar]
- Spang, P.; Herrmann, C.; Roesch, F. Bifunctional gallium-68 chelators: Past, present, and future. Semin. Nucl. Med. 2016, 46, 373–394. [Google Scholar] [CrossRef] [Green Version]
- Sengar, R.S.; Nigam, A.; Geib, S.J.; Wiener, E.C. Syntheses and crystal structures of gadolinium and europium complexes of AAZTA analogues. Polyhedron 2009, 28, 1525–1531. [Google Scholar] [CrossRef]
- Gianolio, E.; Giovenzana, G.B.; Ciampa, A.; Lanzardo, S.; Imperio, D.; Aime, S. A novel method of cellular labeling: Anchoring MR-imaging reporter particles on the outer cell surface. ChemMedChem 2008, 3, 60–62. [Google Scholar] [CrossRef]
- Gugliotta, G.; Botta, M.; Giovenzana, G.B.; Tei, L. Fast and Easy Access to Efficient Bifunctional Chelators for MRI Applications. Bioorg. Med. Chem. Lett. 2009, 19, 3442–3444. [Google Scholar] [CrossRef]
- Gugliotta, G.; Botta, M.; Tei, L. AAZTA-based bifunctional chelating agents for the synthesis of multimeric/dendrimeric MRI contrast agents. Org. Biomol. Chem. 2010, 8, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, J.; Koezle, I.; Bergmann, R.; Tamburini, S.; Bolzati, C.; Tisato, F.; Noll, B.; Klussmann, S.; Vonhoff, S.; Wuest, F.; et al. An 86Y-labeled mirror-image oligonucleotide: Influence of Y-DOTA isomers on the biodistribution in rats. Bioconjug. Chem. 2008, 19, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.M.; Hong, M.K.; Chang, Y.S.; Lee, Y.-S.; Kim, Y.J.; Cheon, G.J.; Lee, D.S.; Chung, J.-K.; Lee, M.C. Preparation of a promising angiogenesis PET imaging agent: 68Ga-Labeled c(RGDyK)–isothiocyanatobenzyl-1,4,7-triazacyclononane-1,4,7-triacetic acid and feasibility studies in mice. J. Nucl. Med. 2008, 49, 830–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Niu, G.; Shi, J.; Liu, S.; Wang, F.; Liu, S.; Chen, X. 68Ga-Labeled cyclic RGD dimers with Gly3 and PEG4 linkers: Promising agents for tumor integrin Avβ3 PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yan, Y.; Liu, S.; Wang, F.; Chen, X. 18F, 64Cu, and 68Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug. Chem. 2009, 20, 1016–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bihan, T.; Navarro, A.-S.; Le Bris, N.; Le Saëc, P.; Gouard, S.; Haddad, F.; Gestin, J.-F.; Chérel, M.; Faivre-Chauvet, A.; Tripier, R. Synthesis of C-functionalized TE1PA and comparison with its analogues. An example of bioconjugation on 9E7.4 MAb for multiple myeloma 64Cu-PET imaging. Org. Biomol. Chem. 2018, 16, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Giesler, R.J.; Erickson, P.W.; Kay, M.S. Enhancing native chemical ligation for challenging chemical protein syntheses. Curr. Opin. Chem. Biol. 2020, 58, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Hawala, I.; De Rosa, L.; Aime, S.; D’Andrea, L.D. An innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach. Chem. Commun. 2020, 56, 3500–3503. [Google Scholar] [CrossRef]
- Ravasco, J.M.J.M.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with maleimides: A useful tool for chemical biology. Chem. Eur. J. 2019, 25, 43–59. [Google Scholar] [CrossRef]
- Bandoli, G.; Dolmella, A.; Tisato, F.; Porchia, M.; Refosco, F. Mononuclear six-coordinated Ga(III) complexes: A comprehensive survey. Coord. Chem. 2009, 253, 56–77. [Google Scholar] [CrossRef]
- Parker, D.; Waldron, B.P. Conformational analysis and synthetic approaches to polydentate perhydro-diazepine ligands for the complexation of gallium(III). Org. Biomol. Chem. 2013, 11, 2827–2838. [Google Scholar] [CrossRef]
- Parker, D.; Waldron, B.P.; Yufit, D.S. Crystallographic and solution NMR structural analyses of four hexacoordinated gallium(III) complexes based on ligands derived from 6-amino-perhydro-1,4-diazepine. Dalton Trans. 2013, 42, 8001–8008. [Google Scholar] [CrossRef]
- Waldron, B.P.; Parker, D.; Burchardt, C.; Yufit, D.S.; Zimny, M.; Roesch, F. Structure and stability of hexadentate complexes of ligands based on AAZTA for efficient PET labelling with gallium-68. Chem. Commun. 2013, 49, 579–581. [Google Scholar] [CrossRef]
- Seemann, J.; Waldron, B.P.; Roesch, F.; Parker, D. Approaching ‘kit-type’ labelling with 68Ga: The DATA chelators. ChemMedChem 2015, 10, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; Nagel, J.; Waldron, B.P.; Parker, D.; Tóth, I.; Brücher, E.; Rösch, F.; Baranyai, Z. Equilibrium, kinetic and structural properties of gallium(III) and some divalent metal complexes formed with the new DATAm and DATA5m ligands. Chem. Eur. J. 2017, 23, 10358–10371. [Google Scholar] [CrossRef] [Green Version]
- Clarke, E.T.; Martell, A.E. Stabilities of trivalent metal ion complexes of the tetraacetate derivatives of 12-, 13- and 14-membered tetraazamacrocycles. Inorg. Chim. Acta 1991, 190, 37–46. [Google Scholar] [CrossRef]
- Tei, L.; Gugliotta, G.; Fekete, M.; Kálmán, F.K.; Botta, M. Mn(II) complexes of novel hexadentate AAZTA-like chelators: A solution thermodynamics and relaxometric study. Dalton Trans. 2011, 40, 2025–2032. [Google Scholar] [CrossRef]
- Greifenstein, L.; Späth, D.; Sinnes, J.P.; Grus, T.; Rösch, F. Mild and efficient 64Cu labeling of perhydro-1,4-diazepine derivatives for potential use with large peptides, proteins and antibodies. Radiochim. Acta 2020, 108, 555–563. [Google Scholar] [CrossRef]
- Klasen, B.; Moon, E.S.; Rösch, F. AAZTA5-Squaramide ester competing with DOTA-, DTPA- and CHX-A″-DTPA-analogues: Promising tool for 177Lu-labeling of monoclonal antibodies under mild conditions. Nucl. Med. Biol. 2021, 96, 80–93. [Google Scholar] [CrossRef]
- Grus, T.; Lahnif, H.; Klasen, B.; Moon, E.-S.; Greifenstein, L.; Roesch, F. Squaric acid-based radiopharmaceuticals for tumor imaging and therapy. Bioconjug. Chem. 2021, 32, 1223–1231. [Google Scholar] [CrossRef]
- Vologdin, N.; Rolla, G.A.; Botta, M.; Tei, L. Orthogonal synthesis of a heterodimeric ligand for the development of the GdIII–GaIII ditopic complex as a potential pH-sensitive MRI/PET probe. Org. Biomol. Chem. 2013, 11, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.P.; Parker, D.; Reany, O.; Aime, S.; Botta, M.; Castellano, G.; Gianolio, E.; Pagliarin, R. pH-Dependent modulation of relaxivity and luminescence in macrocyclic gadolinium and europium complexes based on reversible intramolecular sulfonamide ligation. J. Am. Chem. Soc. 2001, 123, 7601–7609. [Google Scholar] [CrossRef]
- Wu, Z.; Zha, Z.; Choi, S.R.; Plössl, K.; Zhu, L.; Kung, H.F. New 68Ga-phenA bisphosphonates as potential bone imaging agents. Nucl. Med. Biol. 2016, 43, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Calderoni, L.; Farolfi, A.; Pianori, D.; Maietti, E.; Cabitza, V.; Lambertini, A.; Ricci, G.; Telo, S.; Lodi, F.; Castellucci, P.; et al. Evaluation of an automated module synthesis and a sterile cold kit–based preparation of 68Ga-PSMA-11 in patients with prostate cancer. J. Nucl. Med. 2020, 61, 716–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, P.; Lamarca, A.; Navalkissoor, S.; Calero, J.; Chan, P.S.; Julyan, P.; Sierra, M.; Caplin, M.; Valle, J. Safety, tolerability and clinical implementation of ‘ready-to-use’ 68-gallium-DOTA0-Tyr3-Octreotide (68Ga-DOTATOC) (SomaKIT TOC) for injection in patients diagnosed with gastroenteropancreatic neuroendocrine tumours (GEP-NETs). ESMO Open 2020, 5, e000650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khawar, A.; Eppard, E.; Roesch, F.; Ahmadzadehfar, H.; Kürpig, S.; Meisenheimer, M.; Gaertner, F.C.; Essler, M.; Bundschuh, R.A. Preliminary results of biodistribution and dosimetric analysis of [68Ga]Ga-DOTAZOL: A new zoledronate-based bisphosphonate for PET/CT diagnosis of bone diseases. Ann. Nucl. Med. 2019, 33, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Khawar, A.; Eppard, E.; Roesch, F.; Ahmadzadehfar, H.; Kürpig, S.; Meisenheimer, M.; Gaertner, F.C.; Essler, M.; Bundschuh, R.A. Biodistribution and post-therapy dosimetric analysis of [177Lu]Lu-DOTAZOL in patients with osteoblastic metastases: First results. EJNMMI Res. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.; Eppard, E.; Lehnert, W.; Jiménez-Franco, L.D.; Soza-Ried, C.; Ceballos, M.; Ribbeck, J.; Kluge, A.; Rösch, F.; Meckel, M.; et al. Evaluation of safety and dosimetry of 177Lu-DOTA-ZOL for therapy of bone metastases. J. Nucl. Med. 2021, 62, 1126–1132. [Google Scholar] [CrossRef]
- Orteca, G.; Sinnes, J.-P.; Rubagotti, S.; Iori, M.; Capponi, P.C.; Piel, M.; Rösch, F.; Ferrari, E.; Asti, M. Gallium-68 and scandium-44 labelled radiotracers based on curcumin structure linked to bifunctional chelators: Synthesis and characterization of potential PET radiotracers. J. Inorg. Biochem. 2020, 204, 110954. [Google Scholar] [CrossRef]
- Rahbar, K.; Afshar-Oromieh, A.; Jadvar, H.; Ahmadzadehfar, H. PSMA theranostics: Current status and future directions. Mol. Imaging 2018, 17, 153601211877606. [Google Scholar] [CrossRef] [Green Version]
- Sinnes, J.-P.; Bauder-Wüst, U.; Schäfer, M.; Moon, E.S.; Kopka, K.; Rösch, F. 68Ga, 44Sc and 177Lu-labeled AAZTA5-PSMA-617: Synthesis, radiolabeling, stability and cell binding compared to DOTA-PSMA-617 analogues. EJNMMI Radiopharm. Chem. 2020, 5, 28. [Google Scholar] [CrossRef]
- Ghiani, S.; Hawala, I.; Szikra, D.; Trencsényi, G.; Baranyai, Z.; Nagy, G.; Vágner, A.; Stefania, R.; Pandey, S.; Maiocchi, A. Synthesis, radiolabeling, and pre-clinical evaluation of [44Sc]Sc-AAZTA conjugate PSMA inhibitor, a new tracer for high-efficiency imaging of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2351–2362. [Google Scholar] [CrossRef]
- Le Roux, J.; Kleynhans, J.; Rubow, S. The use of HEPES-buffer in the production of gallium-68 radiopharmaceuticals—Time to reconsider strict Pharmacopoeial limits? EJNMMI Radiopharm. Chem. 2021, 6, 15. [Google Scholar] [CrossRef]
- Greifenstein, L.; Grus, T.; Nagel, J.; Sinnes, J.P.; Rösch, F. Synthesis and labeling of a squaric acid containing PSMA-inhibitor coupled to AAZTA5 for versatile labeling with 44Sc, 64Cu, 68Ga and 177Lu. Appl. Radiat. Isot. 2020, 156, 108867. [Google Scholar] [CrossRef]
- Lahnif, H.; Grus, T.; Pektor, S.; Greifenstein, L.; Schreckenberger, M.; Rösch, F. Hybrid chelator-based PSMA radiopharmaceuticals: Translational approach. Molecules 2021, 26, 6332. [Google Scholar] [CrossRef]
- Banerjee, S.; Pillai, M.R.A.; Knapp, F.F. Lutetium-177 therapeutic radiopharmaceuticals: Linking chemistry, radiochemistry, and practical applications. Chem. Rev. 2015, 115, 2934–2974. [Google Scholar] [CrossRef]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Extended structure–activity relationship and pharmacokinetic investigation of (4-quinolinoyl)glycyl-2-cyanopyrrolidine inhibitors of fibroblast activation protein (FAP). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef]
- Lindner, T.; Giesel, F.L.; Kratochwil, C.; Serfling, S.E. Radioligands targeting fibroblast activation protein (FAP). Cancers 2021, 13, 5744. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.S.; Elvas, F.; Vliegen, G.; De Lombaerde, S.; Vangestel, C.; De Bruycker, S.; Bracke, A.; Eppard, E.; Greifenstein, L.; Klasen, B.; et al. Targeting fibroblast activation protein (FAP): Next generation PET radiotracers using squaramide coupled bifunctional DOTA and DATA5m chelators. EJNMMI Radiopharm. Chem. 2020, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, B.; Gärtner, F.C.; Marinova, M.; Attenberger, U.; Meisenheimer, M.; Toma, M.; Kristiansen, G.; Feldmann, G.; Moon, E.S.; Roesch, F.; et al. [68Ga]Ga-DATA5m.SA.FAPi PET/CT: Specific tracer-uptake in focal nodular hyperplasia and potential role in liver tumor imaging. Nuklearmedizin 2020, 59, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.S.; Van Rymenant, Y.; Battan, S.; De Loose, J.; Bracke, A.; Van der Veken, P.; De Meester, I.; Rösch, F. In vitro evaluation of the squaramide-conjugated fibroblast activation protein inhibitor-based agents AAZTA5.SA.FAPi and DOTA.SA.FAPi. Molecules 2021, 26, 3482. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.P.; Breeman, W.A.P.; Kooij, P.P.M.; Lameris, J.S.; Bakker, W.H.; Koper, J.W.; Ausema, L.; Reubi, J.C.; Lamberts, S.W.J. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet 1989, 333, 242–244. [Google Scholar] [CrossRef]
- Bakker, W.H.; Albert, R.; Bruns, C.; Breeman, W.A.P.; Hofland, L.J.; Marbach, P.; Pless, J.; Pralet, D.; Stolz, B.; Koper, J.W.; et al. [111In-DTPA-D-Phe1]-Octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: Synthesis, radiolabeling and In vitro validation. Life Sci. 1991, 49, 1583–1591. [Google Scholar] [CrossRef]
- Otte, A.; Jermann, E.; Behe, M.; Goetze, M.; Bucher, H.C.; Roser, H.W.; Heppeler, A.; Mueller-Brand, J.; Maecke, H.R. DOTATOC: A powerful new tool for receptor-mediated radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 792–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Schmitt, J.S.; Ginj, M.; Mäcke, H.R.; Bernard, B.F.; Krenning, E.; de Jong, M.; Wenger, S.; Reubi, J.-C. DOTA-NOC, a high-affinity ligand of somatostatin receptor subtypes 2, 3 and 5 for labelling with various radiometals. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1338–1347. [Google Scholar] [CrossRef]
- Manzoni, L.; Belvisi, L.; Arosio, D.; Bartolomeo, M.P.; Bianchi, A.; Brioschi, C.; Buonsanti, F.; Cabella, C.; Casagrande, C.; Civera, M.; et al. Synthesis of Gd and 68Ga complexes in conjugation with a conformationally optimized RGD sequence as potential MRI and PET tumor-imaging probes. ChemMedChem 2012, 7, 1084–1093. [Google Scholar] [CrossRef]
- Reubi, J.C. Targeting CCK receptors in human cancers. Curr. Top. Med. Chem. 2007, 7, 1239–1242. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 781–793. [Google Scholar] [CrossRef]
- Klingler, M.; Hörmann, A.A.; Guggenberg, E.V. Cholecystokinin-2 receptor targeting with radiolabeled peptides: Current status and future directions. Curr. Med. Chem. 2020, 27, 7112–7132. [Google Scholar] [CrossRef]
- Pfister, J.; Summer, D.; Rangger, C.; Petrik, M.; von Guggenberg, E.; Minazzi, P.; Giovenzana, G.B.; Aloj, L.; Decristoforo, C. Influence of a novel, versatile bifunctional chelator on theranostic properties of a minigastrin analogue. EJNMMI Res. 2015, 5, 74. [Google Scholar] [CrossRef] [Green Version]
- Tornesello, A.L.; Aurilio, M.; Accardo, A.; Tarallo, L.; Barbieri, A.; Arra, C.; Tesauro, D.; Morelli, G.; Aloj, L. Gastrin and cholecystokinin peptide-based radiopharmaceuticals: An in vivo and in vitro comparison. J. Peptide Sci. 2011, 17, 405–412. [Google Scholar] [CrossRef]
- Moreno, P.; Ramos-Álvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, X.; Liu, H.; Bu, L.; Ma, X.; Cheng, K.; Li, J.; Tian, M.; Zhang, H.; Cheng, Z. A comparative study of radiolabeled bombesin analogs for the PET imaging of prostate cancer. J. Nucl. Med. 2013, 54, 2132–2138. [Google Scholar] [CrossRef] [Green Version]
- Hofstetter, M.; Moon, E.S.; D’Angelo, F.; Geissbühler, L.; Alberts, I.; Afshar-Oromieh, A.; Rösch, F.; Rominger, A.; Gourni, E. Effect of the versatile bifunctional chelator AAZTA5 on the radiometal labelling properties and the In vitro performance of a gastrin releasing peptide receptor antagonist. EJNMMI Radiopharm. Chem. 2020, 5, 29. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of radiolabeled somatostatin analogs for cancer imaging and therapy. Molecules 2020, 25, 4012. [Google Scholar] [CrossRef]
- Bombardieri, E.; Ambrosini, V.; Aktolun, C.; Baum, R.P.; Bishof-Delaloye, A.; Del Vecchio, S.; Maffioli, L.; Mortelmans, L.; Oyen, W.; Pepe, G.; et al. 111In-Pentetreotide scintigraphy: Procedure guidelines for tumour imaging. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1441–1448. [Google Scholar] [CrossRef]
- Sinnes, J.-P.; Nagel, J.; Rösch, F. AAZTA5/AAZTA5-TOC: Synthesis and radiochemical evaluation with 68Ga, 44Sc and 177Lu. EJNMMI Radiopharm. Chem. 2019, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Seemann, J.; Waldron, B.; Parker, D.; Roesch, F. DATATOC: A novel conjugate for kit-type 68Ga labelling of TOC at ambient temperature. EJNMMI Radiopharm. Chem. 2016, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, J.; Eppard, E.; Waldron, B.P.; Ross, T.L.; Roesch, F. Cation exchange-based post-processing of 68Ga-eluate: A comparison of three solvent systems for labelling of DOTATOC, NO2APBP and DATAm. Appl. Radiat. Isot. 2015, 98, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Nagel, J.; Sinnes, J.-P.; Roesch, F.; Maina, T. Novel bifunctional DATA chelator for quick access to site-directed PET 68Ga-radiotracers: Preclinical proof-of-principle with [Tyr3]octreotide. Dalton Trans. 2017, 46, 14584–14590. [Google Scholar] [CrossRef] [PubMed]
- Sinnes, J.-P.; Nagel, J.; Waldron, B.P.; Maina, T.; Nock, B.A.; Bergmann, R.K.; Ullrich, M.; Pietzsch, J.; Bachmann, M.; Baum, R.P.; et al. Instant kit preparation of 68Ga-radiopharmaceuticals via the hybrid chelator DATA: Clinical translation of [68Ga]Ga-DATA-TOC. EJNMMI Res. 2019, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maina, T.; Nock, B.; Nagel, J.; Sinnes, J.P.; Baum, R.; Roesch, F. In vitro binding affinities and initial in vivo evaluation of Ga-68-DATA-TOC. J. Nucl. Med. 2016, 57, 1112. [Google Scholar]
- Gaertner, F.C.; Plum, T.; Kreppel, B.; Eppard, E.; Meisenheimer, M.; Strunk, H.; Bundschuh, R.A.; Sinnes, J.P.; Rösch, F.; Essler, M. Clinical evaluation of [68Ga]Ga-DATA-TOC in comparison to [68Ga]Ga-DOTA-TOC in patients with neuroendocrine tumours. Nucl. Med. Biol. 2019, 76–77, 1–9. [Google Scholar] [CrossRef]
- Yadav, D.; Ballal, S.; Yadav, M.P.; Tripathi, M.; Roesch, F.; Bal, C. Evaluation of [68Ga]Ga-DATA-TOC for imaging of neuroendocrine tumours: Comparison with [68Ga]Ga-DOTA-NOC PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 860–869. [Google Scholar] [CrossRef]
- Giovenzana, G.B.; Palmisano, G.; Sisti, M.; Cavallotti, C.; Aime, S.; Calabi, L. Multidentate Aza Ligands Able to Complex Metal Ions and the Use Thereof in Diagnostics and Therapy. International Patent Application No. WO2003008390A1, 30 January 2003. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fersing, C.; Masurier, N.; Rubira, L.; Deshayes, E.; Lisowski, V. AAZTA-Derived Chelators for the Design of Innovative Radiopharmaceuticals with Theranostic Applications. Pharmaceuticals 2022, 15, 234. https://doi.org/10.3390/ph15020234
Fersing C, Masurier N, Rubira L, Deshayes E, Lisowski V. AAZTA-Derived Chelators for the Design of Innovative Radiopharmaceuticals with Theranostic Applications. Pharmaceuticals. 2022; 15(2):234. https://doi.org/10.3390/ph15020234
Chicago/Turabian StyleFersing, Cyril, Nicolas Masurier, Léa Rubira, Emmanuel Deshayes, and Vincent Lisowski. 2022. "AAZTA-Derived Chelators for the Design of Innovative Radiopharmaceuticals with Theranostic Applications" Pharmaceuticals 15, no. 2: 234. https://doi.org/10.3390/ph15020234