
Mind the Gap—Deciphering GPCR Pharmacology Using 3D Pharmacophores and Artificial Intelligence
 , , , , , ,
, , , , , ,
Abstract
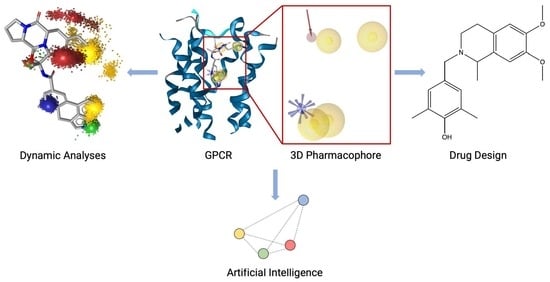
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
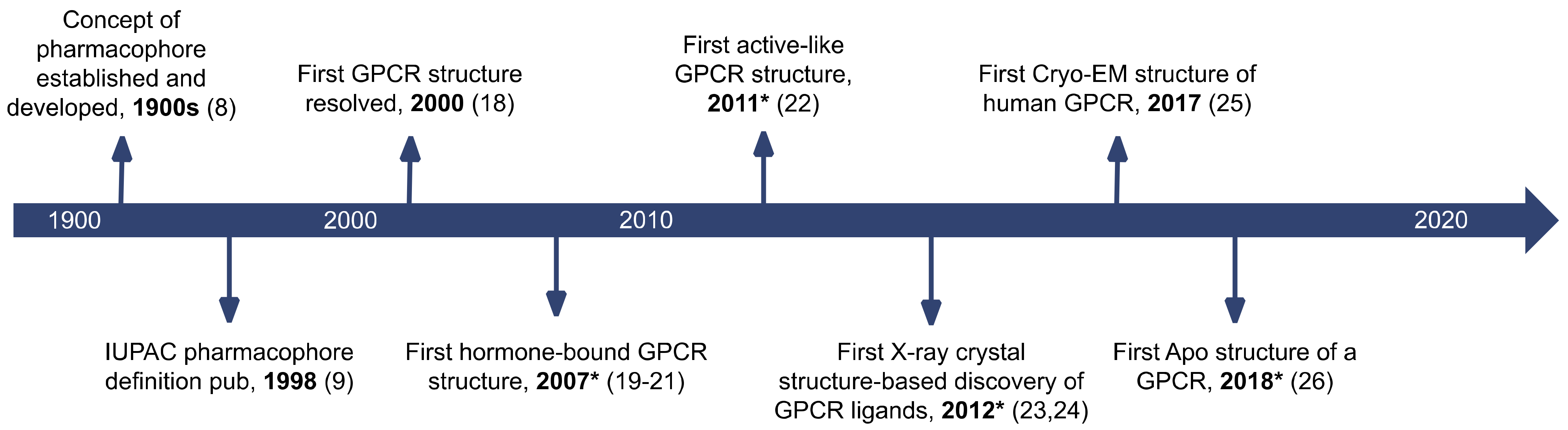
1. Introduction
2. Virtual Screening
2.1. Orthosteric Ligands
2.1.1. Bombesin Receptor 1
2.1.2. Bradykinin Receptors
2.1.3. β3-Adrenergic Receptor
2.1.4. Cannabinoid Receptor 2
2.1.5. G Protein-Coupled Bile Acid Receptor 1
2.1.6. G Protein-Coupled Estrogen Receptor
2.1.7. Histamine H3 Receptor
2.1.8. Melanin-Concentrating Hormone Receptor-1
2.1.9. Neurotensin Receptor Type 1
2.1.10. Protease-Activated Receptor 2
2.2. Medicinal Plants in Virtual Screening
2.2.1. TGR5
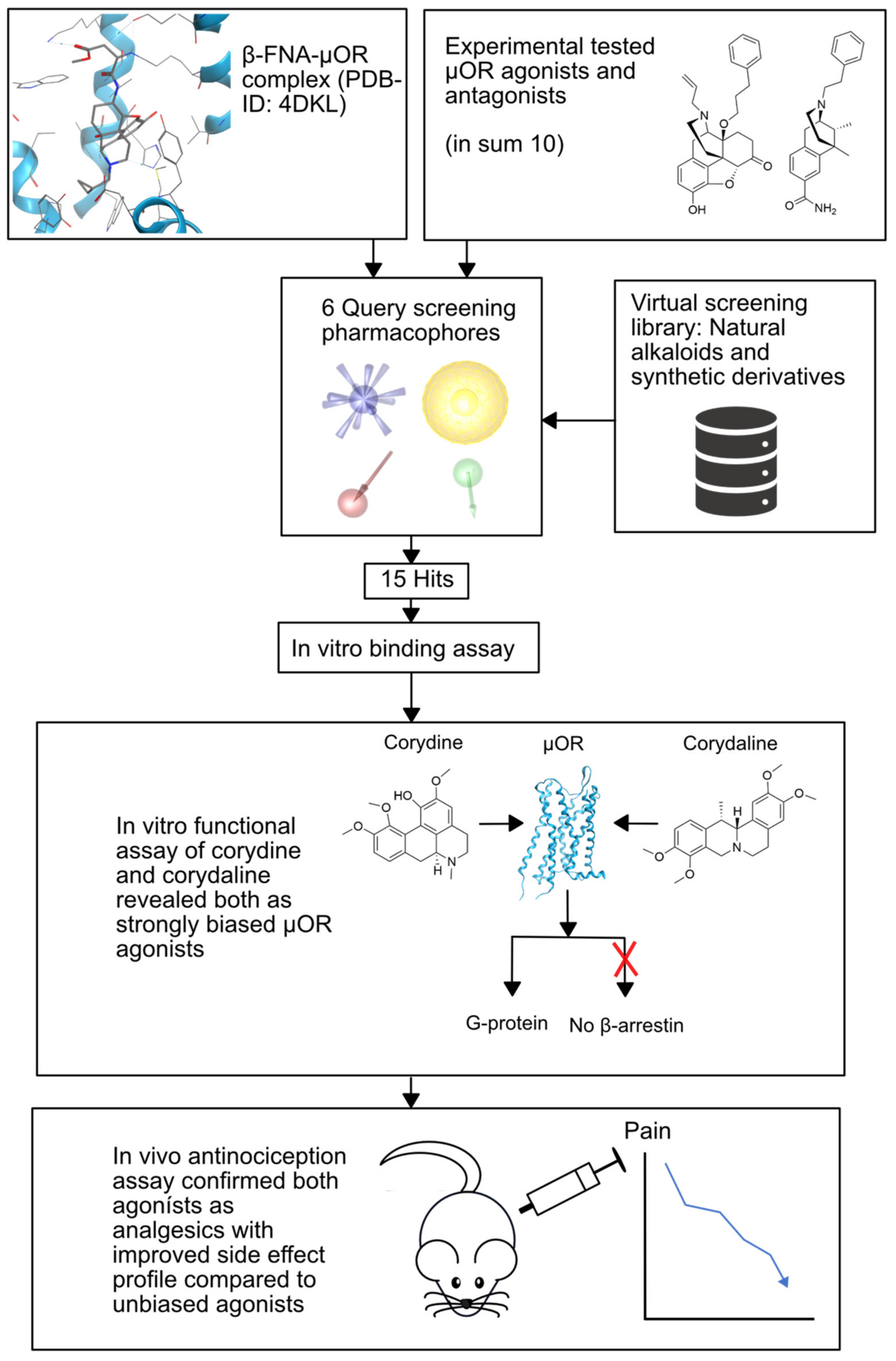
2.2.2. µ-Opioid Receptor
2.3. Biased Ligands
2.3.1. Opioid Receptors
2.4. Allosteric Modulators
2.4.1. Chemokine Receptors
2.4.2. Metabotropic Glutamate Receptor 1
3. Structure-Activity Relationships and QSAR
3.1. Parathyroid Hormone-1 Receptor
3.2. Dopamine D2 Receptor
3.3. Serotonin 5-HT7 Receptor
3.4. TGR5
3.5. G Protein-Coupled Receptor 40
4. Scaffold Hopping and Hit-to-Lead Optimization
4.1. Histamine H4 Receptor
4.2. Somatostatin Receptor Subtype-2
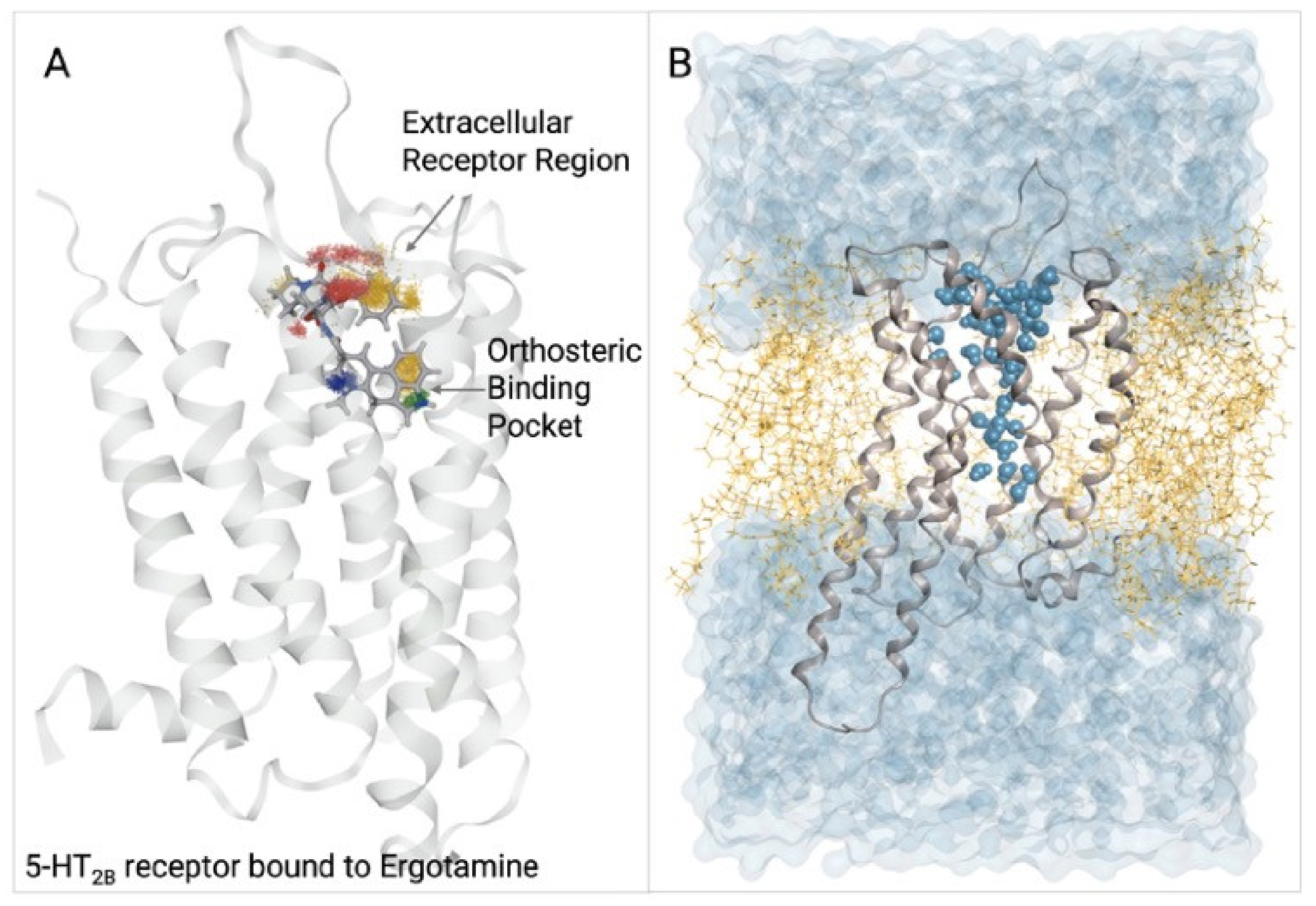
4.3. Serotonin 5-HT2B Receptor
4.4. G Protein-Coupled Receptor 139
5. Dynamics in GPCR-Based Pharmacophore Modeling
5.1. Dynamic Pharmacophores
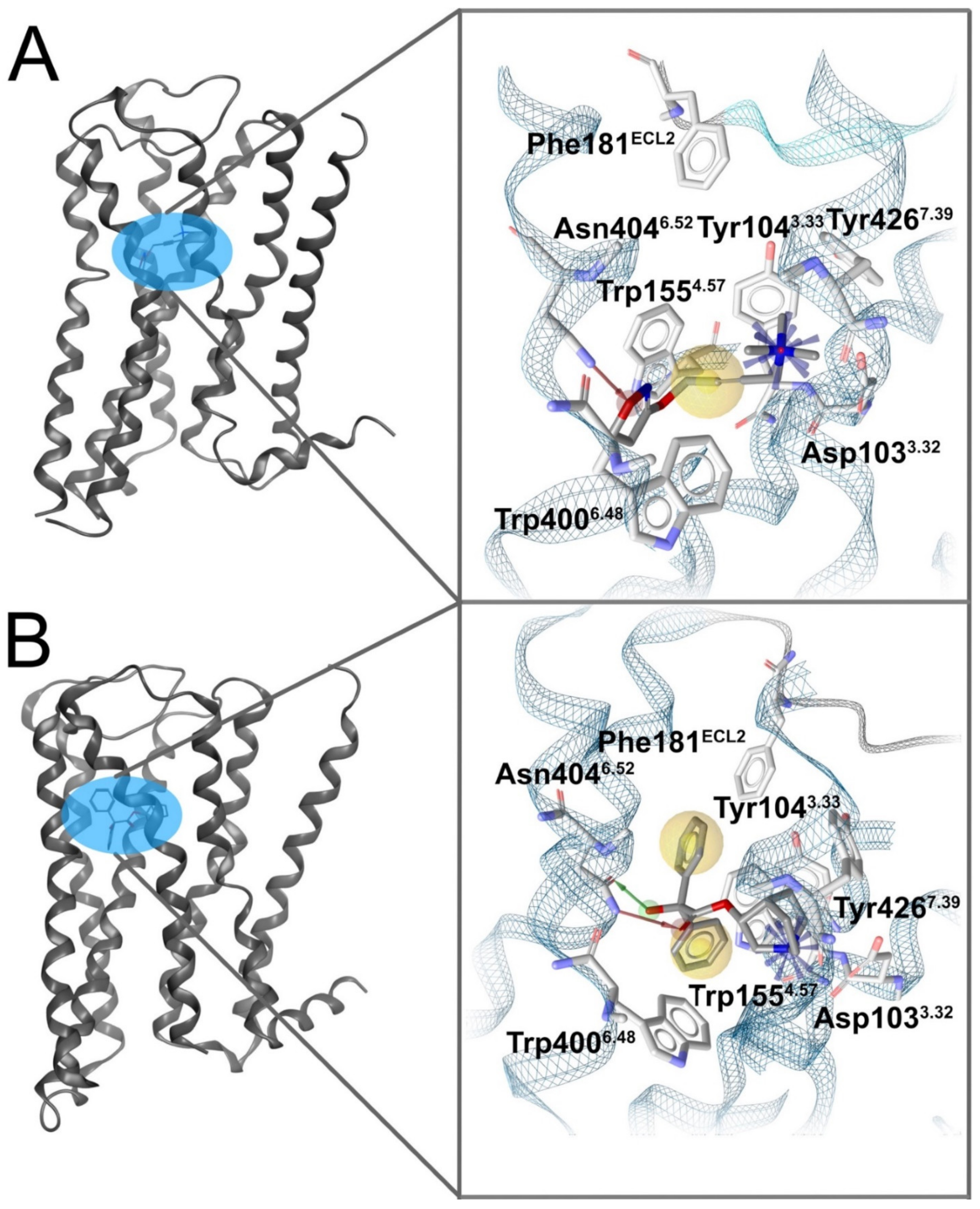
5.1.1. M1 Receptor
5.1.2. 5-HT2B Receptor
5.2. PyRod
5.3. Dopamine D2/D3 Receptor
6. Machine Learning and 3D Pharmacophore Models in GPCR Drug Discovery
7. Discussion
7.1. Challenges: LBDD Is Favored over SBDD in GPCR Research
7.2. Evaluating PBVS and DBVS in GPCR Ligand Discovery
7.3. Comparing Different Studies on the Same Receptor
8. Future Perspectives
De-Orphanizing GPCRs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
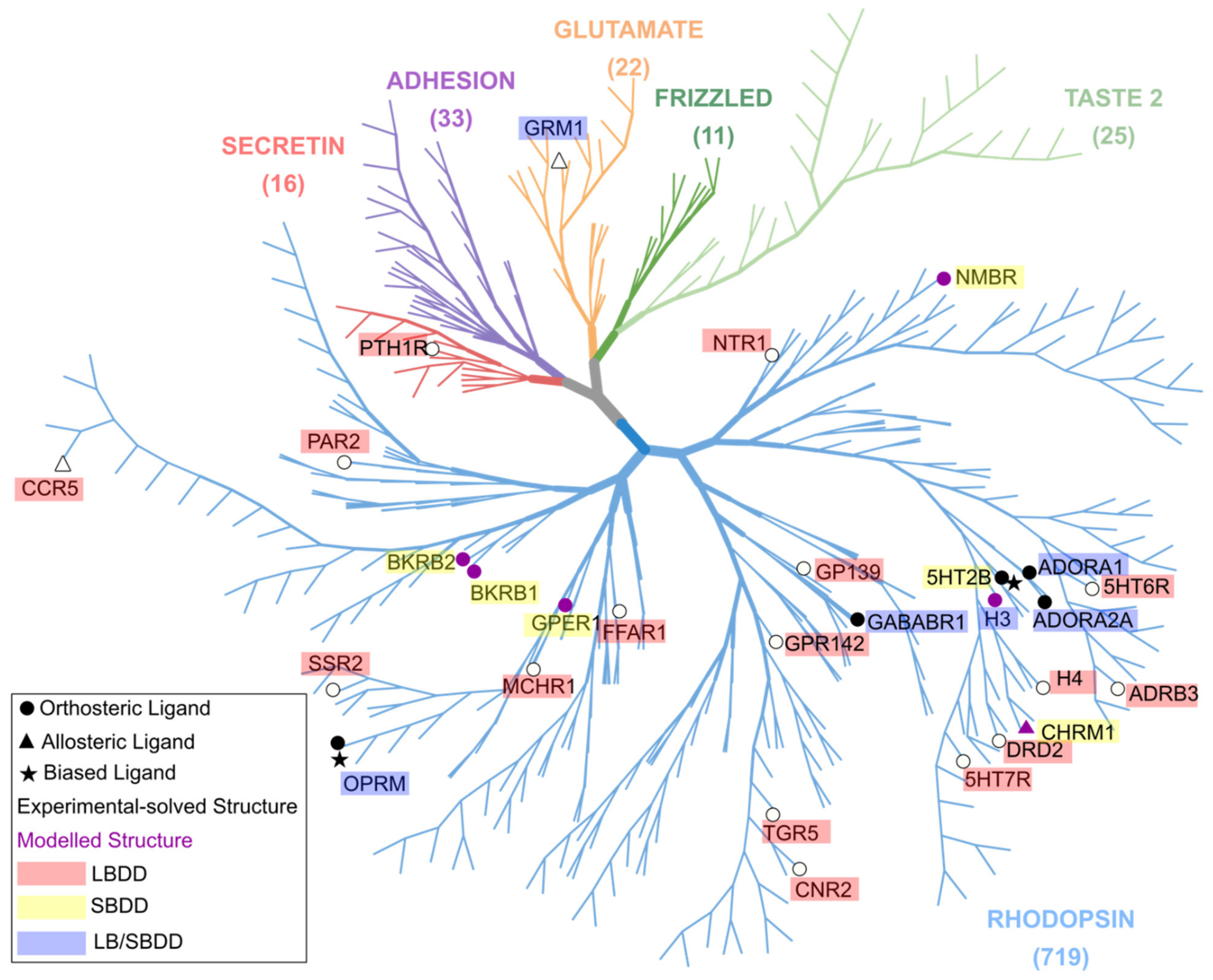
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Bock, A.; Bermudez, M. Allosteric coupling and biased agonism in G protein-coupled receptors. FEBS J. 2021, 288, 2513–2528. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Güner, O.F.; Bowen, J.P. Setting the Record Straight: The Origin of the Pharmacophore Concept. J. Chem. Inf. Model. 2014, 54, 1269–1283. [Google Scholar] [CrossRef]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Sato, T.; Honma, T.; Yokoyama, S. Combining machine learning and pharmacophore-based interaction fingerprint for in silico screening. J. Chem. Inf. Model. 2010, 50, 170–185. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef] [Green Version]
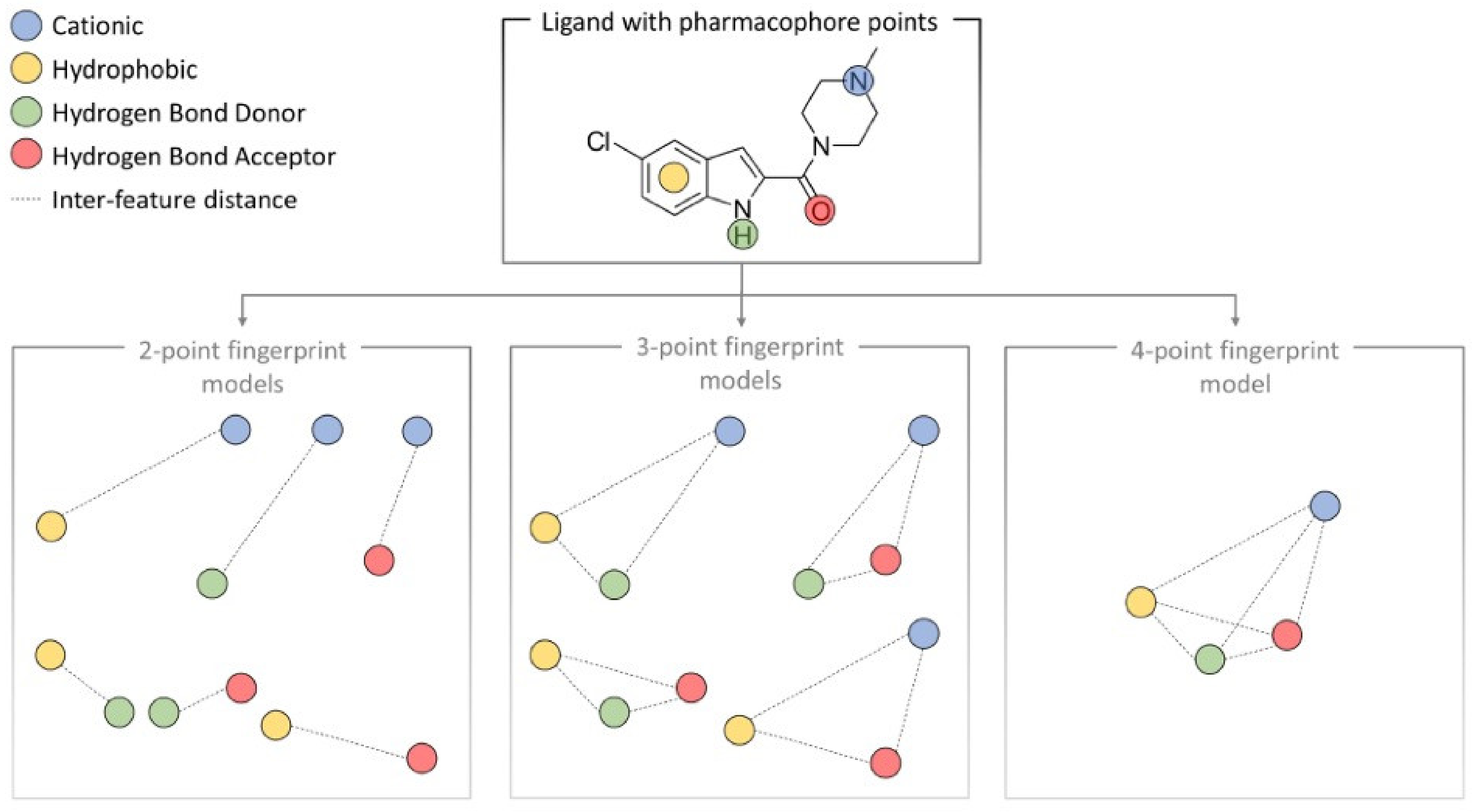
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Black, J.W.; Duncan, W.A.M.; Shanks, R.G. Comparison Of Some Properties Of Pronethalol And Propranolol. Br. J. Pharmacol. Chemother. 1965, 25, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke Craig, A.; Motoshima, H.; Fox Brian, A.; Trong Isolde, L.; Teller David, C.; Okada, T.; Stenkamp Ronald, E.; et al. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen Søren, G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-Resolution Crystal Structure of an Engineered Human β2-Adrenergic G Protein–Coupled Receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.F.; Choi, H.-J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Rosenbaum Daniel, M.; Cherezov, V.; Hanson Michael, A.; Rasmussen Søren, G.F.; Thian Foon, S.; Kobilka Tong, S.; Choi, H.-J.; Yao, X.-J.; Weis William, I.; Stevens Raymond, C.; et al. GPCR Engineering Yields High-Resolution Structural Insights into β2-Adrenergic Receptor Function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.F.; Choi, H.-J.; DeVree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-Triazine Derivatives as Adenosine A2A Antagonists using Structure Based Drug Design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Langmead, C.J.; Andrews, S.P.; Congreve, M.; Errey, J.C.; Hurrell, E.; Marshall, F.H.; Mason, J.S.; Richardson, C.M.; Robertson, N.; Zhukov, A.; et al. Identification of Novel Adenosine A2A Receptor Antagonists by Virtual Screening. J. Med. Chem. 2012, 55, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.-L.; Khoshouei, M.; Radjainia, M.; Zhang, Y.; Glukhova, A.; Tarrasch, J.; Thal, D.M.; Furness, S.G.B.; Christopoulos, G.; Coudrat, T.; et al. Phase-plate cryo-EM structure of a class B GPCR–G-protein complex. Nature 2017, 546, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wu, Y.; Xu, T.-H.; de Waal, P.W.; He, Y.; Pu, M.; Chen, Y.; DeBruine, Z.J.; Zhang, B.; Zaidi, S.A.; et al. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 2018, 560, 666–670. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Homan, E.J.; Wikström, H.V.; Grol, C.J. Molecular modeling of the dopamine D2 and serotonin 5-HT1A receptor binding modes of the enantiomers of 5-OMe-BPAT. Bioorg. Med. Chem. 1999, 7, 1805–1820. [Google Scholar] [CrossRef]
- Chen, X.; Lu, F.; Luo, G.; Ren, Y.; Ma, J.; Zhang, Y. Discovery of selective farnesoid X receptor agonists for the treatment of hyperlipidemia from traditional Chinese medicine based on virtual screening and in vitro validation. J. Biomol. Struct. Dyn. 2020, 38, 4461–4470. [Google Scholar] [CrossRef]
- El-Zohairy, M.A.; Zlotos, D.P.; Berger, M.R.; Adwan, H.H.; Mandour, Y.M. Discovery of Novel CCR5 Ligands as Anticolorectal Cancer Agents by Sequential Virtual Screening. ACS Omega 2021, 6, 10921–10935. [Google Scholar] [CrossRef]
- Ghamari, N.; Zarei, O.; Reiner, D.; Dastmalchi, S.; Stark, H.; Hamzeh-Mivehroud, M. Histamine H3 receptor ligands by hybrid virtual screening, docking, molecular dynamics simulations, and investigation of their biological effects. Chem. Biol. Drug Des. 2019, 93, 832–843. [Google Scholar] [CrossRef]
- Hu, J.; Feng, Z.; Ma, S.; Zhang, Y.; Tong, Q.; Alqarni, M.H.; Gou, X.; Xie, X.-Q. Difference and Influence of Inactive and Active States of Cannabinoid Receptor Subtype CB2: From Conformation to Drug Discovery. J. Chem. Inf. Model. 2016, 56, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Kaserer, T.; Lantero, A.; Schmidhammer, H.; Spetea, M.; Schuster, D. μ Opioid receptor: Novel antagonists and structural modeling. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kaserer, T.; Steinacher, T.; Kainhofer, R.; Erli, F.; Sturm, S.; Waltenberger, B.; Schuster, D.; Spetea, M. Identification and characterization of plant-derived alkaloids, corydine and corydaline, as novel mu opioid receptor agonists. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Ko, K.; Kim, H.-J.; Ho, P.-S.; Lee, S.O.; Lee, J.-E.; Min, C.-R.; Kim, Y.C.; Yoon, J.-H.; Park, E.-J.; Kwon, Y.-J.; et al. Discovery of a Novel Highly Selective Histamine H4 Receptor Antagonist for the Treatment of Atopic Dermatitis. J. Med. Chem. 2018, 61, 2949–2961. [Google Scholar] [CrossRef]
- Wang, J.; Shu, M.; Wang, Y.; Hu, Y.; Wang, Y.; Luo, Y.; Lin, Z. Identification of potential CCR5 inhibitors through pharmacophore-based virtual screening, molecular dynamics simulation and binding free energy analysis. Mol. Biosyst. 2016, 12, 3396–3406. [Google Scholar] [CrossRef]
- Evenseth, L.M.; Warszycki, D.; Bojarski, A.J.; Gabrielsen, M.; Sylte, I. In silico methods for the discovery of orthosteric GABAB receptor compounds. Molecules 2019, 24, 935. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.W.; Cho, N.C.; Min, S.J.; Cho, Y.S.; Park, K.D.; Seo, S.H.; No, K.T.; Pae, A.N. Novel Scaffold Identification of mGlu1 Receptor Negative Allosteric Modulators Using a Hierarchical Virtual Screening Approach. Chem. Biol. Drug Des. 2016, 87, 239–256. [Google Scholar] [CrossRef]
- Helal, M.A.; Chittiboyina, A.G.; Avery, M.A. Identification of a new small molecule chemotype of Melanin Concentrating Hormone Receptor-1 antagonists using pharmacophore-based virtual screening. Bioorg. Med. Chem. Lett. 2019, 29, 126741. [Google Scholar] [CrossRef]
- Kaushik, A.C.; Kumar, S.; Wei, D.Q.; Sahi, S. Structure based virtual screening studies to identify novel potential compounds for GPR142 and their relative dynamic analysis for study of type 2 diabetes. Front. Chem. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabeen, A.; Vijayram, R.; Ranganathan, S. A two-stage computational approach to predict novel ligands for a chemosensory receptor. Curr. Res. Struc. Biol. 2020, 2, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, X.; Peng, W.; Wang, L.; Ye, W.; Zhao, Y.; Yin, W.; Chen, W.-D.; Li, W.; Wang, Y.-D. Ligand-based pharmacophore modeling, virtual screening and biological evaluation to identify novel TGR5 agonists. RSC Adv. 2021, 11, 9403–9409. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, A.; Sondergard, C.; Sweeney, P.; Arnatt, C.K. A series of indole-thiazole derivatives act as GPER agonists and inhibit breast cancer cell growth. ACS Med. Chem. Lett. 2018, 9, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.-C.; Seo, S.-H.; Kim, D.; Shin, J.-S.; Ju, J.; Seong, J.; Seo, S.H.; Lee, I.; Lee, K.-T.; Kim, Y.K. Pharmacophore-based virtual screening, biological evaluation and binding mode analysis of a novel protease-activated receptor 2 antagonist. J. Comput. Aided Mol. Des. 2016, 30, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.X.; Li, G.H.; Gao, Y.D.; Huang, J.F. Pharmacophore-Map-Pick: A Method to Generate Pharmacophore Models for All Human GPCRs. Mol. Inform. 2016, 35, 81–91. [Google Scholar] [CrossRef]
- Kirchweger, B.; Kratz, J.M.; Ladurner, A.; Grienke, U.; Langer, T.; Dirsch, V.M.; Rollinger, J.M. In silico workflow for the discovery of natural products activating the G protein-coupled bile acid receptor 1. Front. Chem. 2018, 6, 242. [Google Scholar] [CrossRef]
- Shiri, F.; Teymoori, M. In silico approaches to explore structure of new GPR 119 agonists for treatment of type 2 diabetes mellitus. Med. Chem. Res. 2017, 26, 947–961. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, K.; Li, X.-D.; Zhang, D.-L.; Xu, F. Discovery of novel antagonists of human neurotensin receptor 1 on the basis of ligand and protein structure. Biomed. Pharmacother. 2016, 84, 147–157. [Google Scholar] [CrossRef]
- Nath, V.; Ahuja, R.; Kumar, V. Identification of novel G-protein-coupled receptor 40 (GPR40) agonists by hybrid in silico-screening techniques and molecular dynamics simulations thereof. J. Biomol. Struct. Dyn. 2019, 37, 3764–3787. [Google Scholar] [CrossRef]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B1 receptor antagonists binding. J. Mol. Graph. Model. 2016, 68, 184–196. [Google Scholar] [CrossRef] [Green Version]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B2 receptor antagonists binding. J. Comput. Aided. Mol. Des. 2016, 30, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals 2020, 13, 197. [Google Scholar] [CrossRef]
- Sindhu, T.; Srinivasan, P. Pharmacophore modeling, comprehensive 3D-QSAR, and binding mode analysis of TGR5 agonists. J. Recept Signal Transduct. Res. 2017, 37, 109–123. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Kao, R.Y.; Shapiro, R. Virtual screening to enrich hit lists from high-throughput screening: A case study on small-molecule inhibitors of angiogenin. Proteins Struct. Funct. Genet. 2003, 50, 81–93. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Mol. Model. Proteins 2008, 433, 365–382. [Google Scholar]
- Jaiteh, M.; Rodríguez-Espigares, I.; Selent, J.; Carlsson, J. Performance of virtual screening against GPCR homology models: Impact of template selection and treatment of binding site plasticity. PLoS Comput. Biol. 2020, 16, e1007680. [Google Scholar] [CrossRef] [Green Version]
- Moffat, K.; Gillet, V.J.; Whittle, M.; Bravi, G.; Leach, A.R. A comparison of field-based similarity searching methods. J. Chem. Inf. Model. 2008, 48, 719–729. [Google Scholar] [CrossRef]
- Hähnke, V.; Todoroff, N.; Rodrigues, T.; Schneider, G. Significance estimation for sequence-based chemical similarity searching (PhAST) and application to AuroraA kinase inhibitors. Future Med. Chem. 2012, 4, 1897–1906. [Google Scholar] [CrossRef]
- Braga, R.C.; Andrade, C.H. Assessing the performance of 3D pharmacophore models in virtual screening: How good are they? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.-T.; Pitis, P.M.; Gotchev, D.; Yuan, C. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Scopton, A.P.; Rives, M.-L.; Bikbulatov, R.V.; Polepally, P.R.; Brown, P.J.; Kenakin, T.; Javitch, J.A.; Zjawiony, J.K.; Roth, B.L. Identification of novel functionally selective κ-opioid receptor scaffolds. Mol. Pharmacol. 2014, 85, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M. Distinct β-arrestin-and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragle, L.E.; Palanisamy, D.J.; Joe, M.J.; Stein, R.S.; Norman, D.D.; Tigyi, G.; Baker, D.L.; Parrill, A.L. Discovery and synthetic optimization of a novel scaffold for hydrophobic tunnel-targeted autotaxin inhibition. Bioorg. Med. Chem. 2016, 24, 4660–4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Edwards, P.C.; Burghammer, M.; Villa, C.; Schertler, G.F. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004, 343, 1409–1438. [Google Scholar] [CrossRef]
- Ujiantari, N.S.O.; Ham, S.; Nagiri, C.; Shihoya, W.; Nureki, O.; Hutchinson, D.S.; Schuster, D. Pharmacophore-guided Virtual Screening to Identify New β3-adrenergic Receptor Agonists. Mol. Inform. 2021, 41, 2100223. [Google Scholar] [CrossRef]
- Specs. Available online: www.specs.net (accessed on 18 July 2022).
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Takaharu, M.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 197, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Mao, C.; Guo, L.; Lin, J.; Ming, Q.; Xiao, P.; Wu, X.; Shen, Q.; Guo, S.; Shen, D.-D. Structural basis of GPBAR activation and bile acid recognition. Nature 2020, 587, 499–504. [Google Scholar] [CrossRef]
- Gemkow, M.J.; Davenport, A.J.; Harich, S.; Ellenbroek, B.A.; Cesura, A.; Hallett, D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov. Today 2009, 14, 509–515. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. Zinc–A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Frandsen, I.O.; Boesgaard, M.W.; Fidom, K.; Hauser, A.S.; Isberg, V.; Bräuner-Obsorne, H.; Wellendorph, P.; Gloriam, D.E. Identification of Histamine H3 Receptor Ligands Using a New Crystal Structure Fragment-based Method. Sci. Rep. 2017, 7, 4829. [Google Scholar] [CrossRef] [Green Version]
- Munk, C.; Isberg, V.; Mordalski, S.; Harpsøe, K.; Rataj, K.; Hauser, A.S.; Kolb, P.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: The G protein–coupled receptor database–An introduction. Br. J. Pharmacol. 2016, 173, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Sabetghadam, A.; Grabowiecka-Nowak, A.; Kania, A.; Gugula, A.; Blasiak, E.; Blasiak, T.; Ma, S.; Gundlach, A.L.; Blasiak, A. Melanin-concentrating hormone and orexin systems in rat nucleus incertus: Dual innervation, bidirectional effects on neuron activity, and differential influences on arousal and feeding. Neuropharmacology 2018, 139, 238–256. [Google Scholar] [CrossRef]
- Kawata, Y.; Okuda, S.; Hotta, N.; Igawa, H.; Takahashi, M.; Ikoma, M.; Kasai, S.; Ando, A.; Satomi, Y.; Nishida, M. A novel and selective melanin-concentrating hormone receptor 1 antagonist ameliorates obesity and hepatic steatosis in diet-induced obese rodent models. Eur. J. Pharmacol. 2017, 796, 45–53. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Huo, X.-K.; Liu, J.; Yu, Z.-L.; Wang, Y.-F.; Wang, C.; Tian, X.-G.; Ning, J.; Feng, L.; Sun, C.-P.; Zhang, B.-J. Alisma orientale extract exerts the reversing cholestasis effect by activation of farnesoid X receptor. Phytomedicine 2018, 42, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Amar, M.J.; Guo, G.; Brewer, H.B.; Gonzalez, F.J.; Sinal, C.J. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 2003, 278, 2563–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef]
- Carino, A.; Biagioli, M.; Marchianò, S.; Scarpelli, P.; Zampella, A.; Limongelli, V.; Fiorucci, S. Disruption of TFGβ-SMAD3 pathway by the nuclear receptor SHP mediates the antifibrotic activities of BAR704, a novel highly selective FXR ligand. Pharmacol. Res. 2018, 131, 17–31. [Google Scholar] [CrossRef]
- Spetea, M.; Faheem Asim, M.; Wolber, G.; Schmidhammer, H. The μ opioid receptor and ligands acting at the μ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Hung. Acta Physiol. 2008, 95, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Schmidhammer, H.; Spetea, M. Synthesis of 14-alkoxymorphinan derivatives and their pharmacological actions. Top. Curr. Chem. 2011, 299, 63–91. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Puls, K.; Olivé-Marti, A.-L.; Pach, S.; Pinter, B.; Erli, F.; Wolber, G.; Spetea, M. In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist. Pharmaceuticals 2022, 15, 680. [Google Scholar] [CrossRef]
- Jacobson, M.L.; Browne, C.A.; Lucki, I. Kappa Opioid Receptor Antagonists as Potential Therapeutics for Stress-Related Disorders. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 615–636. [Google Scholar] [CrossRef]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Ehlert, F.J. Quantifying GPCR allostery and biased signaling. In GPCRs; Jastrzebska, B., Park, P.S.H., Eds.; Academic Press: London, UK, 2020; pp. 143–169. [Google Scholar]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef]
- Kolb, P.; Kenakin, T.; Alexander, S.P.H.; Bermudez, M.; Bohn, L.M.; Breinholt, C.S.; Bouvier, M.; Hill, S.J.; Kostenis, E.; Martemyanov, K.A.; et al. Community guidelines for GPCR ligand bias: IUPHAR review 32. Br. J. Pharmacol. 2022, 179, 3651–3674. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.E.; Kelly, E. A Biased View of μ-Opioid Receptors? Mol. Pharmacol. 2019, 96, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tong, J.; Ding, K.; Zhou, Q.; Zhao, S. GPCR Allosteric Modulator Discovery. Protein Allostery Drug Discov. 2019, 1163, 225–251. [Google Scholar]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2019, 62, 88–127. [Google Scholar] [CrossRef]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [Green Version]
- Pervaiz, A.; Zepp, M.; Mahmood, S.; Ali, D.M.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc: A potential therapeutic option for metastatic breast cancer. Cell. Oncol. 2019, 42, 93–106. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Li, X.; Zhu, D.; Zhao, N.; Yao, H.; Lin, K. Characteristics of parathyroid hormone-1 receptor agonists and antagonists. Future Med. Chem. 2019, 11, 817–831. [Google Scholar] [CrossRef]
- Salmas, R.E.; Stein, M.; Yurtsever, M.; Seeman, P.; Erol, I.; Mestanoglu, M.; Durdagi, S. The signaling pathway of dopamine D2 receptor (D2R) activation using normal mode analysis (NMA) and the construction of pharmacophore models for D2R ligands. J. Biomol. Struct. Dyn. 2017, 35, 2040–2048. [Google Scholar] [CrossRef]
- Shi, L.; Javitch, J.A. The binding site of aminergic G protein-coupled receptors: The transmembrane segments and second extracellular loop. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 437. [Google Scholar] [CrossRef]
- Surgand, J.S.; Rodrigo, J.; Kellenberger, E.; Rognan, D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins 2006, 62, 509–538. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Kuhne, S.; De Esch, I.; Leurs, R.; De Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Free, R.B.; Barnaeva, E.; Conroy, J.L.; Doyle, T.; Miller, B.; Bryant-Genevier, M.; Taylor, M.K.; Hu, X.; Dulcey, A.E.; et al. Discovery, optimization, and characterization of novel D2 dopamine receptor selective antagonists. J. Med. Chem. 2014, 57, 3450–3463. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Zuk, J.; Matosiuk, D. Comparative molecular field analysis and molecular dynamics studies of the dopamine D2 receptor antagonists without a protonatable nitrogen atom. Med. Chem. Res. 2018, 27, 1149–1166. [Google Scholar] [CrossRef] [Green Version]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Kelemen, Á.A.; Satała, G.; Bojarski, A.J.; Keserű, G.M. Spiro [pyrrolidine-3,3′-oxindoles] as 5-HT7 receptor ligands. Bioorg. Med. Chem. Lett. 2018, 28, 2418–2421. [Google Scholar] [CrossRef]
- López-Rodríguez, M.L.; Porras, E.; Morcillo, M.J.; Benhamú, B.; Soto, L.J.; Lavandera, J.L.; Ramos, J.A.; Olivella, M.; Campillo, M.; Pardo, L. Optimization of the pharmacophore model for 5-HT7R antagonism. Design and synthesis of new naphtholactam and naphthosultam derivatives. J. Med. Chem. 2003, 46, 5638–5650. [Google Scholar] [CrossRef]
- Lopez-Rodríguez, M.A.L.; Porras, E.; Benhamú, B.; Ramos, J.A.; Morcillo, M.J.; Lavandera, J.L. First pharmacophoric hypothesis for 5-HT7 antagonism. Bioorg. Med. Chem. Lett. 2000, 10, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.A.; Sallander, J.; Benhamú, B.; Porras, E.; Campillo, M.; Pardo, L.; Lopez-Rodriguez, M.L. Synthesis of new serotonin 5-HT7 receptor ligands. Determinants of 5-HT7/5-HT1A receptor selectivity. J. Med. Chem. 2009, 52, 2384–2392. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ishida, A.; Tajina, Y.; Okabe, Y.; Matshushita, T.; Sekiguchi, T.; Imaide, S.; Aoki, S.; Nishio, T.; Komagata, T.; Iwaki, M.; et al. Discovery and SAR Studies of Orally Active Somatostatin Receptor Subtype-2 (SSTR2) Agonists for the Treatment of Acromegaly. ACS Chem. Neurosci. 2020, 11, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; Abdel-Raziq, M.S. Pharmacophore-based tailoring of biphenyl amide derivatives as selective 5-hydroxytryptamine 2B receptor antagonists. Med. Chem. Commun. 2018, 9, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ma, J.; Lin, X.; Huang, X.-P.; Wu, K.; Huang, N. Structure-Based Discovery of Novel and Selective 5-Hydroxytryptamine 2B Receptor Antagonists for the Treatment of Irritable Bowel Syndrome. J. Med. Chem. 2016, 59, 707–720. [Google Scholar] [CrossRef]
- Moss, N.; Choi, Y.; Cogan, D.; Flegg, A.; Kahrs, A.; Loke, P.; Meyn, O.; Nagaraja, R.; Napier, S.; Parker, A.; et al. A new class of 5-HT 2B antagonists possesses favorable potency, selectivity, and rat pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2009, 19, 2206–2210. [Google Scholar] [CrossRef]
- Shehata, M.A.; Nøhr, A.C.; Lissa, D.; Bisig, C.; Isberg, V.; Andersen, K.B.; Harpsøe, K.; Björkling, F.; Bräuner-Osborne, H.; Gloriam, D.E. Novel Agonist Bioisosteres and Common Structure-Activity Relationships for The Orphan G Protein-Coupled Receptor GPR139. Sci. Rep. 2016, 6, 36681. [Google Scholar] [CrossRef] [Green Version]
- Ferruz, N.; Doerr, S.; Vanase-Frawley, M.A.; Zou, Y.; Chen, X.; Marr, E.S.; Nelson, R.T.; Kormos, B.L.; Wager, T.T.; Hou, X.; et al. Dopamine D3 receptor antagonist reveals a cryptic pocket in aminergic GPCRs. Sci. Rep. 2018, 8, 897. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.; Hunenberger, P.H.; Bakowies, D.; Baron, R.; Burgi, R.; Geerke, D.P.; Heinz, T.N.; Kastenholz, M.A.; Krautler, V.; Oostenbrink, C.; et al. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Riniker, S. Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview. J. Chem. Inf. Model. 2018, 58, 565–578. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Espigares, I.; Torrens-Fontanals, M.; Tiemann, J.K.S.; Aranda-Garcia, D.; Ramirez-Anguita, J.M.; Stepniewski, T.M.; Worp, N.; Varela-Rial, A.; Morales-Pastor, A.; Medel-Lacruz, B.; et al. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 2020, 17, 777–787. [Google Scholar] [CrossRef]
- Mortier, J.; Prevost, J.R.C.; Sydow, D.; Teuchert, S.; Omieczynski, C.; Bermudez, M.; Frederick, R.; Wolber, G. Arginase Structure and Inhibition: Catalytic Site Plasticity Reveals New Modulation Possibilities. Sci. Rep. 2017, 7, 13616. [Google Scholar] [CrossRef] [Green Version]
- Volpato, D.; Kauk, M.; Messerer, R.; Bermudez, M.; Wolber, G.; Bock, A.; Hoffmann, C.; Holzgrabe, U. The Role of Orthosteric Building Blocks of Bitopic Ligands for Muscarinic M1 Receptors. ACS Omega 2020, 5, 31706–31715. [Google Scholar] [CrossRef]
- Denzinger, K.; Nguyen, T.N.; Noonan, T.; Wolber, G.; Bermudez, M. Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors. Int. J. Mol. Sci. 2020, 21, 9728. [Google Scholar] [CrossRef]
- Schaller, D.; Pach, S.; Wolber, G. PyRod: Tracing Water Molecules in Molecular Dynamics Simulations. J. Chem. Inf. Model. 2019, 59, 2818–2829. [Google Scholar] [CrossRef] [PubMed]
- Schaller, D.; Wolber, G. PyRod Enables Rational Homology Model-based Virtual Screening Against MCHR1. Mol. Inform. 2020, 39, e2000020. [Google Scholar] [CrossRef]
- Jabeen, A.; Ranganathan, S. Applications of machine learning in GPCR bioactive ligand discovery. Curr. Opin. Struct. Biol. 2019, 55, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S.; Kaufman, B. Machine learning and AI-based approaches for bioactive ligand discovery and GPCR-ligand recognition. Methods 2020, 180, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S. Automated discovery of GPCR bioactive ligands. Curr. Opin. Struct. Biol. 2019, 55, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillari, C.; Marcou, G.; Rognan, D. Hot-spots-guided receptor-based pharmacophores (HS-Pharm): A knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J. Chem. Inf. Model. 2008, 48, 1396–1410. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; Fabritiis, G.d. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [Green Version]
- Bento, P.A.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-K. Pharmacophore Perception, Development and Use in Drug Design. Edited by Osman F. Güner. Molecules 2000, 5, 987–989. [Google Scholar] [CrossRef]
- Gobbi, A.; Poppinger, D. Genetic optimization of combinatorial libraries. Biotechnol. Bioeng. 1998, 61, 47–54. [Google Scholar] [CrossRef]
- Wood, D.J.; de Vlieg, J.; Wagener, M.; Ritschel, T. Pharmacophore fingerprint-based approach to binding site subpocket similarity and its application to bioisostere replacement. J. Chem. Inf. Model. 2012, 52, 2031–2043. [Google Scholar] [CrossRef]
- Warszycki, D.; Struski, Ł.; Śmieja, M.; Kafel, R.; Kurczab, R. Pharmacoprint: A Combination of a Pharmacophore Fingerprint and Artificial Intelligence as a Tool for Computer-Aided Drug Design. J. Chem. Inf. Model. 2021, 61, 5054–5065. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, M.; Li, Y.; Hong, Z.; Li, D.; Lin, J. Identification of new potent A1 adenosine receptor antagonists using a multistage virtual screening approach. Eur. J. Med. Chem. 2020, 187, 111936. [Google Scholar] [CrossRef]
- Wang, M.; Hou, S.; Wei, Y.; Li, D.; Lin, J. Discovery of novel dual adenosine A1/A2A receptor antagonists using deep learning, pharmacophore modeling and molecular docking. PLoS Comput. Biol. 2021, 17, e1008821. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wüthrich, K. The GPCR Network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: Hoboken, NJ, USA, 1990. [Google Scholar]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular Docking Screening Using Agonist-Bound GPCR Structures: Probing the A 2A Adenosine Receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castleman, P.; Szabowski, G.; Bowman, D.; Cole, J.; Parrill, A.L.; Baker, D.L. Ligand-based G Protein Coupled Receptor pharmacophore modeling: Assessing the role of ligand function in model development. J. Mol. Graph. Model. 2022, 111, 108107. [Google Scholar] [CrossRef]
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 2010, 7, 221–228. [Google Scholar] [CrossRef]
- Miszta, P.; Pasznik, P.; Jakowiecki, J.; Sztyler, A.; Latek, D.; Filipek, S. GPCRM: A homology modeling web service with triple membrane-fitted quality assessment of GPCR models. Nucleic Acids Res. 2018, 46, W387–W395. [Google Scholar] [CrossRef] [Green Version]
- Worth, C.L.; Kreuchwig A Fau-Kleinau, G.; Kleinau G Fau-Krause, G.; Krause, G. GPCR-SSFE: A comprehensive database of G-protein-coupled receptor template predictions and homology models. BMC Bioinform. 2011, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Ngo, T.; Kufareva, I.; Coleman, J.L.; Graham, R.M.; Abagyan, R.; Smith, N.J. Identifying ligands at orphan GPCRs: Current status using structure-based approaches. Br. J. Pharmacol. 2016, 173, 2934–2951. [Google Scholar] [CrossRef] [Green Version]
- Lemer, C.M.; Rooman Mj Fau-Wodak, S.J.; Wodak, S.J. Protein structure prediction by threading methods: Evaluation of current techniques. Proteins 1995, 23, 337–355. [Google Scholar] [CrossRef]
- Hardin, C.; Pogorelov, T.V.; Luthey-Schulten, Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 2002, 12, 176–181. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Niswnder, C.M.; Katritch, V.; Meiler, J.; Cherezov, V.; et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 2014, 344, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Ballante, F.; Kooistra, A.J.; Kampen, S.; de Graaf, C.; Carlsson, J. Structure-Based Virtual Screening for Ligands of G Protein–Coupled Receptors: What Can Molecular Docking Do for You? Pharmacol. Rev. 2021, 73, 1698. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2005, 20, 13384–13421. [Google Scholar] [CrossRef] [Green Version]
- Pillaiyar, T.; Köse, M.; Namasivayam, V.; Sylvester, K.; Borges, G.; Thimm, D.; von Kügelgen, I.; Müller, C.E. 6-(Ar)Alkylamino-Substituted Uracil Derivatives: Lipid Mimetics with Potent Activity at the Orphan G Protein-Coupled Receptor 84 (GPR84). ACS Omega 2018, 3, 3365–3383. [Google Scholar] [CrossRef]
- Ngan, C.-H.; Hall, D.R.; Zerbe, B.; Grove, L.E.; Kozakov, D.; Vajda, S. FTSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2012, 28, 286–287. [Google Scholar] [CrossRef] [Green Version]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noonan, T.; Denzinger, K.; Talagayev, V.; Chen, Y.; Puls, K.; Wolf, C.A.; Liu, S.; Nguyen, T.N.; Wolber, G. Mind the Gap—Deciphering GPCR Pharmacology Using 3D Pharmacophores and Artificial Intelligence. Pharmaceuticals 2022, 15, 1304. https://doi.org/10.3390/ph15111304
Noonan T, Denzinger K, Talagayev V, Chen Y, Puls K, Wolf CA, Liu S, Nguyen TN, Wolber G. Mind the Gap—Deciphering GPCR Pharmacology Using 3D Pharmacophores and Artificial Intelligence. Pharmaceuticals. 2022; 15(11):1304. https://doi.org/10.3390/ph15111304
Chicago/Turabian StyleNoonan, Theresa, Katrin Denzinger, Valerij Talagayev, Yu Chen, Kristina Puls, Clemens Alexander Wolf, Sijie Liu, Trung Ngoc Nguyen, and Gerhard Wolber. 2022. "Mind the Gap—Deciphering GPCR Pharmacology Using 3D Pharmacophores and Artificial Intelligence" Pharmaceuticals 15, no. 11: 1304. https://doi.org/10.3390/ph15111304