
Kinsenoside Alleviates 17α-Ethinylestradiol-Induced Cholestatic Liver Injury in Rats by Inhibiting Inflammatory Responses and Regulating FXR-Mediated Bile Acid Homeostasis

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. KD Protects against EE-Induced Cholestatic Liver Injury in Rats
2.2. KD Normalizes Cholestasis-Related Serum Biochemical Factors
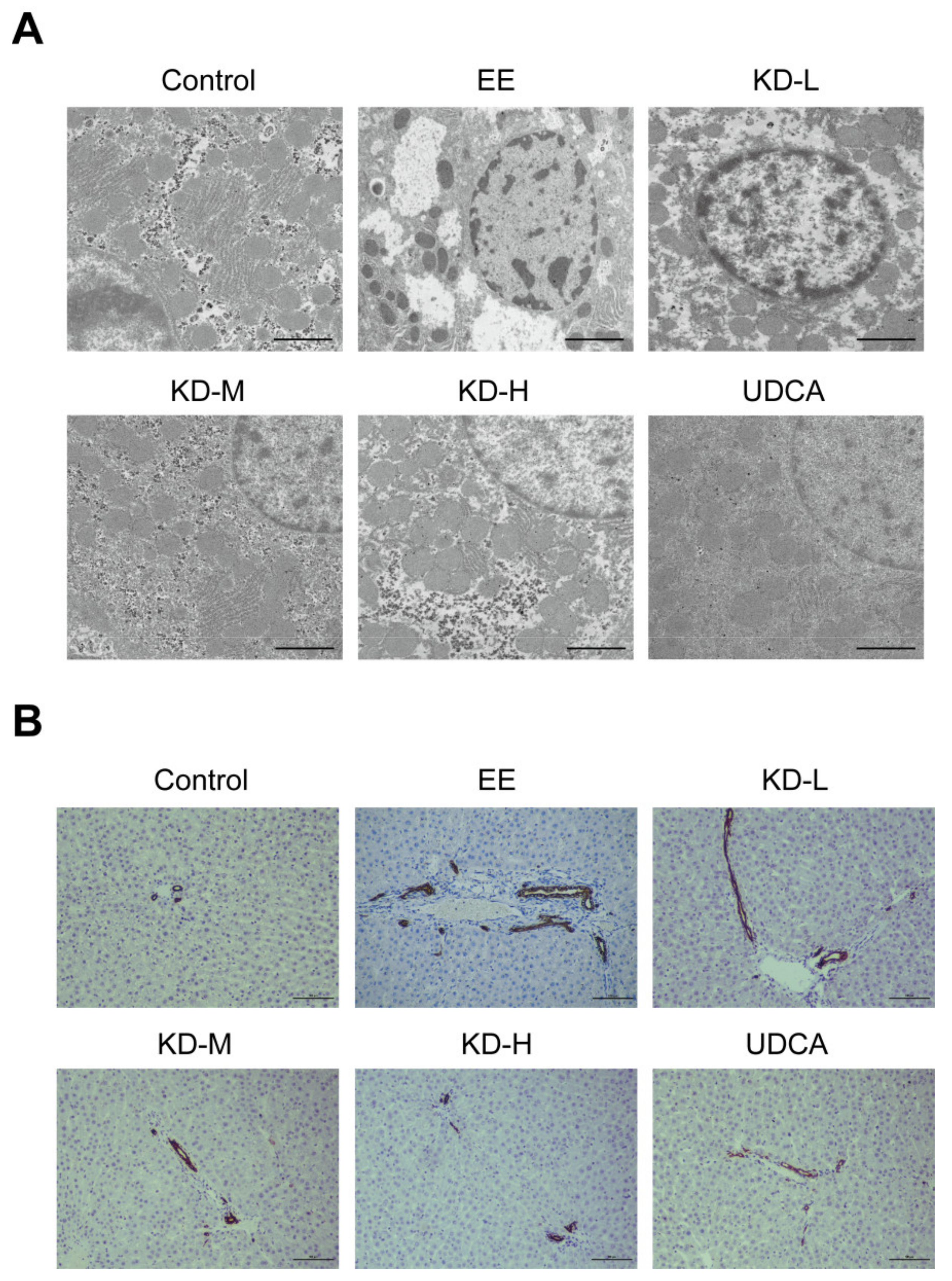
2.3. KD Inhibits Hepatocellular Microstructural Disorder and Bile Duct Cell Proliferation
2.4. KD Reduces the Expression of Pro-Inflammatory Cytokines Caused by EE in the Liver
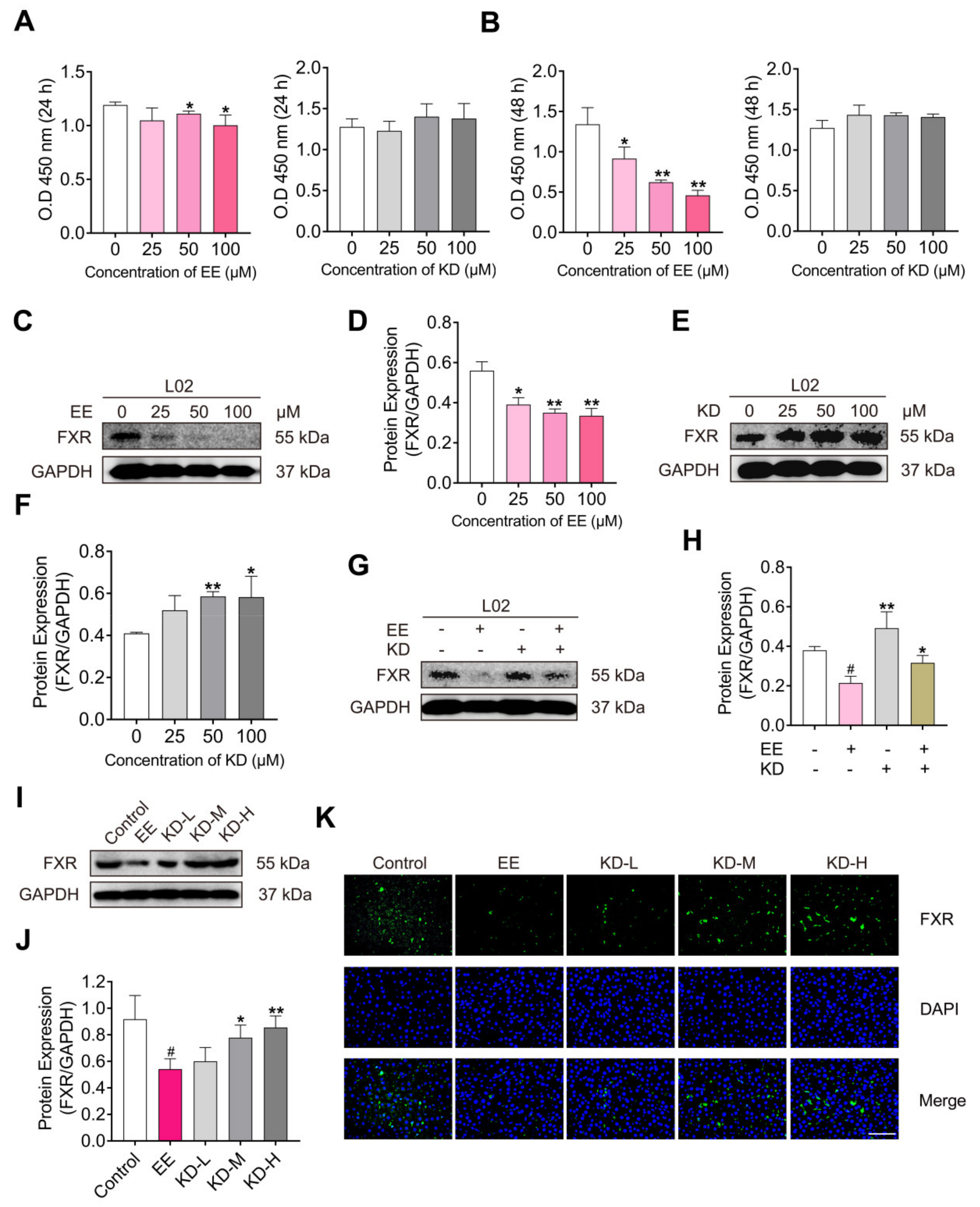
2.5. KD Suppresses EE-Mediated Decrease in FXR In Vitro and In Vivo
2.6. KD Alters the Expression of Transporters, Synthetic and Metabolic Enzyme Involved in Bile Acid Homeostasis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals and Treatments
4.3. Histopathology Assay
4.4. Serum Biochemistry Assay
4.5. Transmission Electron Microscopy
4.6. Immunohistochemistry Staining Assay
4.7. Cell Line and Cell Culture
4.8. Cell Viability Assay
4.9. Western Blotting
4.10. Immunofluorescence Staining Assay
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayappa, M.; Kumar, P.; Goyal, J.P. Prolonged cholestasis after acute viral hepatitis: Successfully treated with oral steroid. BMJ Case Rep. 2020, 13, e234430. [Google Scholar] [CrossRef]
- Chalifoux, S.L.; Konyn, P.G.; Choi, G.; Saab, S. Extrahepatic Manifestations of Primary Biliary Cholangitis. Gut Liver 2017, 11, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Alpert, L.; Hart, J. The Pathology of Alcoholic Liver Disease. Clin. Liver Dis. 2016, 20, 473–489. [Google Scholar] [CrossRef]
- Shipovskaya, A.A.; Dudanova, O.P. Intrahepatic cholestasis in nonalcoholic fatty liver disease. Ter. Arkhiv 2018, 90, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bhamidimarri, K.R.; Schiff, E. Drug-Induced Cholestasis. Clin. Liver Dis. 2013, 17, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.J.; Simon, F.R. Estrogen-Induced Cholestasis: Clues to Pathogenesis and Treatment. Hepatology 2007, 3, 607–613. [Google Scholar] [CrossRef]
- Pasmant, E.; Goussard, P.; Baranes, L.; Laurendeau, I.; Quentin, S.; Ponsot, P.; Consigny, Y.; Farges, O.; Condat, B.; Vidaud, D.; et al. First description of ABCB4 gene deletions in familial low phospholipid-associated chole-lithiasis and oral contraceptives-induced cholestasis. Eur. J. Hum. Genet. 2012, 20, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.; Geenes, V. Intrahepatic Cholestasis of Pregnancy. Obstet. Gynecol. 2014, 124, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihma, V.; Ropponen, A.; Aittomäki, K.; Ylikorkala, O.; Tikkanen, M.J. Postmenopausal estrogen therapy and serum estradiol fatty acid esters in women with and without previous intrahepatic cholestasis of pregnancy. Ann. Med. 2004, 36, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kontturi, M.; Sotaniemi, E. Effect of oestrogen on liver function of prostatic cancer patients. BMJ 1969, 4, 204–205. [Google Scholar] [CrossRef] [Green Version]
- Jansen, P.L.; Ghallab, A.; Vartak, N.; Reif, R.; Schaap, F.G.; Hampe, J.; Hengstler, J.G. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017, 65, 722–738. [Google Scholar] [CrossRef] [Green Version]
- De Vries, E.; Beuers, U. Management of cholestatic disease in 2017. Liver Int. 2017, 37 (Suppl. 1), 123–129. [Google Scholar] [CrossRef] [Green Version]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. Fibrates and cholestasis. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Floreani, A.; Mangini, C. Primary biliary cholangitis: Old and novel therapy. Eur. J. Intern. Med. 2018, 47, 1–5. [Google Scholar] [CrossRef]
- Markham, A.; Keam, S.J. Obeticholic Acid: First Global Approval. Drugs 2016, 76, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Moore, R.; Hess, H.A.; Guo, G.L.; Gonzalez, F.J.; Korach, K.S.; Maronpot, R.R.; Negishi, M. Estrogen Receptor α Mediates 17α-Ethynylestradiol Causing Hepatotoxicity. J. Biol. Chem. 2006, 281, 16625–16631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriquez-Hernandez, L.A.; Flores-Morales, A.; Santana-Farré, R.; Axelson, M.; Nilsson, P.; Norstedt, G.; Fernandez-Perez, L. Role of Pituitary Hormones on 17α-Ethinylestradiol-Induced Cholestasis in Rat. J. Pharmacol. Exp. Ther. 2006, 320, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Xiang, D.; Xiang, D.; He, W.; Liu, Y.; Lan, L.; Li, G.; Jiang, C.; Ren, X.; Liu, D.; et al. Baicalin Protects Against 17alpha-Ethinylestradiol-Induced Cholestasis via the Sirtuin 1/Hepatic Nuclear Receptor-1alpha/Farnesoid X Receptor Pathway. Front. Pharmacol. 2019, 10, 1685. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Nuclear receptors in bile acid metabolism. Drug Metab. Rev. 2013, 45, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Mencarelli, A.; Distrutti, E.; Zampella, A. Farnesoid X receptor: From medicinal chemistry to clinical applications. Future Med. Chem. 2012, 4, 877–891. [Google Scholar] [CrossRef]
- Yuan, Z.Q.; Li, K.W. Role of farnesoid X receptor in cholestasis. J. Dig. Dis. 2016, 17, 501–509. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear Receptors and the Regulation of Drug-Metabolizing Enzymes and Drug Transporters: Implications for Interindividual Variability in Response to Drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Luo, L.; Yu, L.; Chen, X.; Sun, L.; Wang, T.; Hylemon, P.B.; Zhou, H.; Jiang, Z.; et al. Role of AMP-activated protein kinase α1 in 17α-ethinylestradiol-induced cholestasis in rats. Arch. Toxicol. 2016, 91, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Shao, Q.; Zhang, A. Anoectochilus roxburghii: A review of its phytochemistry, pharmacology, and clinical applications. J. Ethnopharmacol. 2017, 209, 184–202. [Google Scholar] [CrossRef]
- Cheng, K.T.; Wang, Y.S.; Chou, H.C.; Chang, C.C.; Lee, C.K.; Juan, S.H. Kinsenoside-mediated lipolysis through an AMPK-dependent pathway in C3H10T1/2 adipocytes: Roles of AMPK and PPARalpha in the lipolytic effect of kinsenoside. Phytomedicine 2015, 22, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.-B.; Hsieh, C.-C.; Wu, J.-B.; Lin, H.; Lin, W.-C. Kinsenoside inhibits the inflammatory mediator release in a type-II collagen induced arthritis mouse model by regulating the T cells responses. BMC Complement. Altern. Med. 2016, 16, 80. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.L.; Liu, Q.; Xiao, B.; Zhou, J.; Zhang, J.G.; Li, Y. The vascular protective properties of kinsenoside isolated from An-oectochilus roxburghii under high glucose condition. Fitoterapia 2013, 86, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.B.; Lin, W.L.; Hsieh, C.C.; Ho, H.Y.; Tsay, H.S.; Lin, W.C. The hepatoprotective activity of kinsenoside from Anoectochilus formosanus. Phytother. Res. 2007, 21, 58–61. [Google Scholar] [CrossRef]
- Hsieh, W.-T.; Tsai, C.-T.; Wu, J.-B.; Hsiao, H.-B.; Yang, L.-C.; Lin, W.-C. Kinsenoside, a high yielding constituent from Anoectochilus formosanus, inhibits carbon tetrachloride induced Kupffer cells mediated liver damage. J. Ethnopharmacol. 2011, 135, 440–449. [Google Scholar] [CrossRef]
- Li, X.; Yuan, Z.; Liu, R.; Hassan, H.M.; Yang, H.; Sun, R.; Zhang, L.; Jiang, Z. UDCA and CDCA alleviate 17α-ethinylestradiol-induced cholestasis through PKA-AMPK pathways in rats. Toxicol. Appl. Pharmacol. 2016, 311, 12–25. [Google Scholar] [CrossRef]
- Zhuo, J.-Y.; Lu, D.; Tan, W.-Y.; Zheng, S.-S.; Shen, Y.-Q.; Xu, X. CK19-positive Hepatocellular Carcinoma is a Characteristic Subtype. J. Cancer 2020, 11, 5069–5077. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y.; Ji, H.; Li, Y. Anti-inflammatory, anti-oxidative stress and novel therapeutic targets for cholestatic liver injury. Biosci. Trends 2019, 13, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-kappaB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Telbisz, Á.M.; Homolya, L. Recent advances in the exploration of the bile salt export pump (BSEP/ABCB11) function. Expert Opin. Ther. Targets 2015, 20, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Liu, L.; Shan, W.; Kong, L.; Chen, N.; Lou, Y.; Zeng, S. The Role of the Sodium-taurocholate Co-transporting Poly-peptide (NTCP) and Bile Salt Export Pump (BSEP) in Related Liver Disease. Curr. Drug Metab. 2019, 20, 377–389. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, P.; Scheinberg, A.R.; Levy, C. Cholestatic liver diseases: New targets, new therapies. Ther. Adv. Gastroenterol. 2018, 11, 1756284818787400. [Google Scholar] [CrossRef]
- Boyer, J.L. New perspectives for the treatment of cholestasis: Lessons from basic science applied clinically. J. Hepatol. 2007, 46, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Yang, R.; Wang, J.; Hu, D.D.; Li, F. PPARalpha activation protects against cholestatic liver injury. Sci. Rep. 2017, 7, 9967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roma, M.G.; Toledo, F.D.; Boaglio, A.C.; Basiglio, C.L.; Crocenzi, F.A.; Sanchez Pozzi, E.J. Ursodeoxycholic acid in cho-lestasis: Linking action mechanisms to therapeutic applications. Clin. Sci. 2011, 121, 523–544. [Google Scholar] [CrossRef] [Green Version]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C.; Parés, A.; et al. Efficacy of Obeticholic Acid in Patients with Primary Biliary Cirrhosis and Inadequate Response to Ursodeoxycholic Acid. Gastroenterology 2015, 148, 751–761.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Jiang, Y.; Zhang, W.; Wang, J.; Wang, R.; Wang, L.; Wei, S.; Wen, J.; Li, H.; Zhao, Y. Natural products for the prevention and treatment of cholestasis: A review. Phytother. Res. 2020, 34, 1291–1309. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Bing, W.; Di, Y.; Hua, L.; Shi-He, L.; Yu-Hua, Z.; Xiu-Guo, H.; Yu-Gang, W.; Qi-Ming, F.; Shih-Mo, Y.; et al. Kinsenoside screening with a microfluidic chip attenuates gouty arthritis through inactivating NF-kappaB signaling in mac-rophages and protecting endothelial cells. Cell. Death Dis. 2016, 7, e2350. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Liu, T.; Tan, W.; Ren, H.; Li, H.; Liu, J.; Cao, H.; Cheng, Q.; Liu, X.; Zhu, H.; et al. Effects of kinsenoside, a potential immunosuppressive drug for autoimmune hepatitis, on dendritic cells/CD8+T cells communication in mice. Hepatology 2016, 64, 2135–2150. [Google Scholar] [CrossRef]
- Biberoglu, E.; Kırbaş, A.; Daglar, K.; Kara, O.; Karabulut, E.; Yakut, H.I.; Danişman, N. Role of inflammation in intrahepatic cholestasis of pregnancy. J. Obstet. Gynaecol. Res. 2016, 42, 252–257. [Google Scholar] [CrossRef]
- Hsiao, H.-B.; Wu, J.-B.; Lin, H.; Lin, W.-C. Kinsenoside Isolated from Anoectochilus Formosanus Suppresses LPS-Stimulated Inflammatory Reactions in Macrophages and Endotoxin Shock in Mice. Shock 2011, 35, 184–190. [Google Scholar] [CrossRef]
- Zhou, F.; Mei, J.; Han, X.; Li, H.; Yang, S.; Wang, M.; Chu, L.; Qiao, H.; Tang, T. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-kappaB/MAPK signaling and protecting chondrocytes. Acta Pharm. Sin. B 2019, 9, 973–985. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, R.; Wang, Z.; Luo, L.; Wu, J.; Zhang, C.; Liu, M.; Shi, C.; Zhou, Y. Kinsenoside ameliorates intervertebral disc degeneration through the activation of AKT-ERK1/2-Nrf2 signaling pathway. Aging 2019, 11, 7961–7977. [Google Scholar] [CrossRef]
- Liu, Q.; Qiao, A.-M.; Yi, L.-T.; Liu, Z.-L.; Sheng, S.-M. Protection of kinsenoside against AGEs-induced endothelial dysfunction in human umbilical vein endothelial cells. Life Sci. 2016, 162, 102–107. [Google Scholar] [CrossRef]
- Luo, X.; Gu, S.; Zhang, Y.; Zhang, J. Kinsenoside Ameliorates Oxidative Stress-Induced RPE Cell Apoptosis and Inhibits An-giogenesis via Erk/p38/NF-kappaB/VEGF Signaling. Front. Pharmacol. 2018, 9, 240. [Google Scholar] [CrossRef]
- Qi, C.-X.; Zhou, Q.; Yuan, Z.; Luo, Z.-W.; Dai, C.; Zhu, H.-C.; Chen, C.-M.; Xue, Y.-B.; Wang, J.-P.; Wang, Y.-F.; et al. Kinsenoside: A Promising Bioactive Compound from Anoectochilus Species. Curr. Med. Sci. 2018, 38, 11–18. [Google Scholar] [CrossRef]
- Lindor, K.D. Farnesoid X receptor agonists for primary biliary cirrhosis. Curr. Opin. Gastroenterol. 2011, 27, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Dröge, C.; Häussinger, D. Targeting FXR in Cholestasis. Organotypic Models Drug Dev. 2019, 256, 299–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, Y.; Lai, Y.; Tang, F.; Luo, Z.; Xue, Y.; Yao, G.; Zhang, Y.; Zhang, J. Efficient Synthesis of Kinsenoside and Goodyeroside A by a Chemo-Enzymatic Approach. Molecules 2014, 19, 16950–16958. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ming, J.; Xu, Q.; Gao, L.; Deng, Y.; Yin, J.; Zhou, Q.; Tong, Q.; Zhang, Y. Kinsenoside Alleviates 17α-Ethinylestradiol-Induced Cholestatic Liver Injury in Rats by Inhibiting Inflammatory Responses and Regulating FXR-Mediated Bile Acid Homeostasis. Pharmaceuticals 2021, 14, 452. https://doi.org/10.3390/ph14050452
Ming J, Xu Q, Gao L, Deng Y, Yin J, Zhou Q, Tong Q, Zhang Y. Kinsenoside Alleviates 17α-Ethinylestradiol-Induced Cholestatic Liver Injury in Rats by Inhibiting Inflammatory Responses and Regulating FXR-Mediated Bile Acid Homeostasis. Pharmaceuticals. 2021; 14(5):452. https://doi.org/10.3390/ph14050452
Chicago/Turabian StyleMing, Jiaxiong, Qianqian Xu, Limin Gao, Yanfang Deng, Jie Yin, Qun Zhou, Qingyi Tong, and Yonghui Zhang. 2021. "Kinsenoside Alleviates 17α-Ethinylestradiol-Induced Cholestatic Liver Injury in Rats by Inhibiting Inflammatory Responses and Regulating FXR-Mediated Bile Acid Homeostasis" Pharmaceuticals 14, no. 5: 452. https://doi.org/10.3390/ph14050452