
P-Glycoprotein (ABCB1/MDR1) and BCRP (ABCG2) Limit Brain Accumulation and Cytochrome P450-3A (CYP3A) Restricts Oral Exposure of the RET Inhibitor Selpercatinib (RETEVMO)

 , , and
, , and
Abstract
:
1. Introduction
2. Results
2.1. In Vitro Transport of Selpercatinib
2.2. Impact of ABCB1, ABCG2 and SLCO1A/1B on Selpercatinib Plasma Pharmacokinetics and Tissue Disposition
2.3. In Vitro Uptake of Selpercatinib
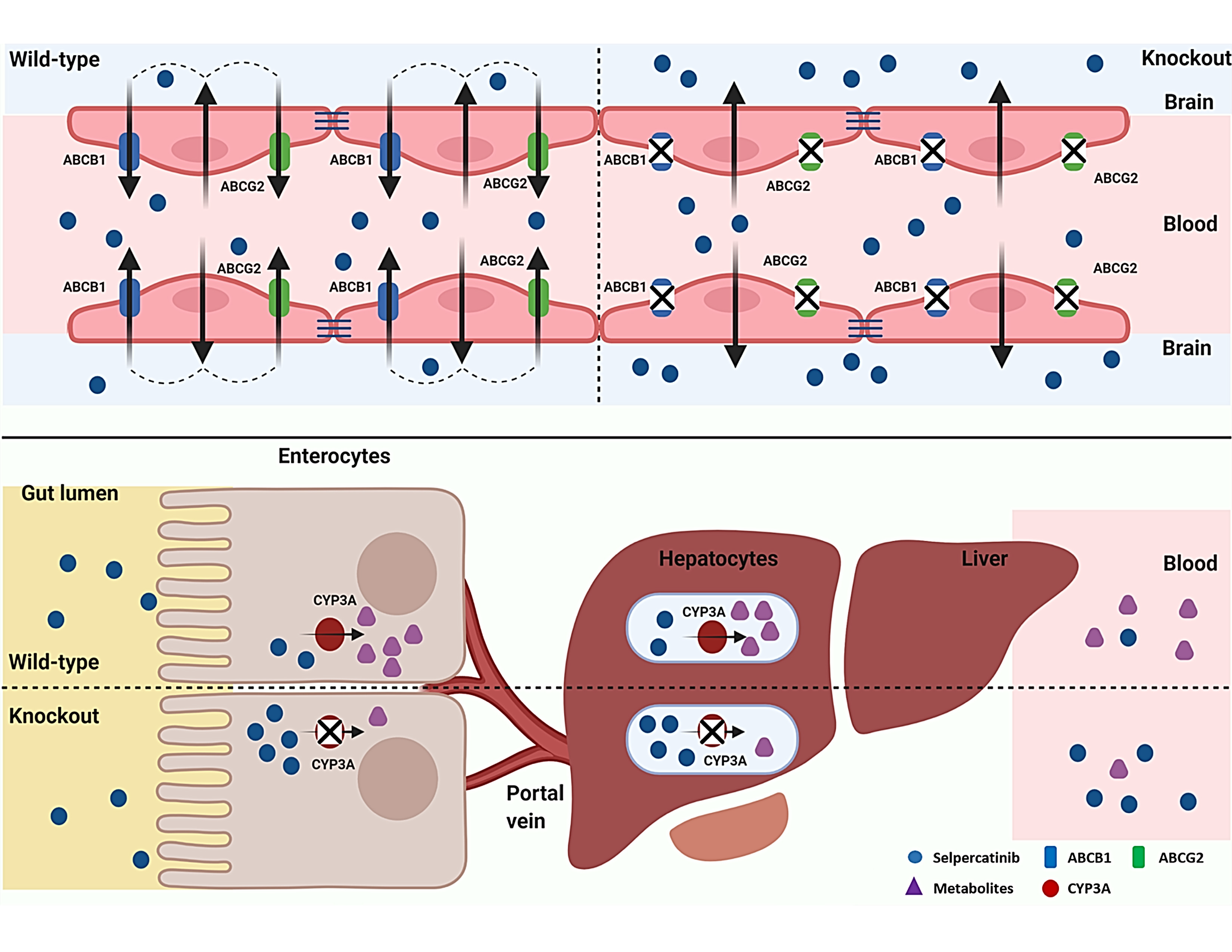
2.4. ABCB1 and ABCG2 Limit Selpercatinib Brain and Testis Exposure
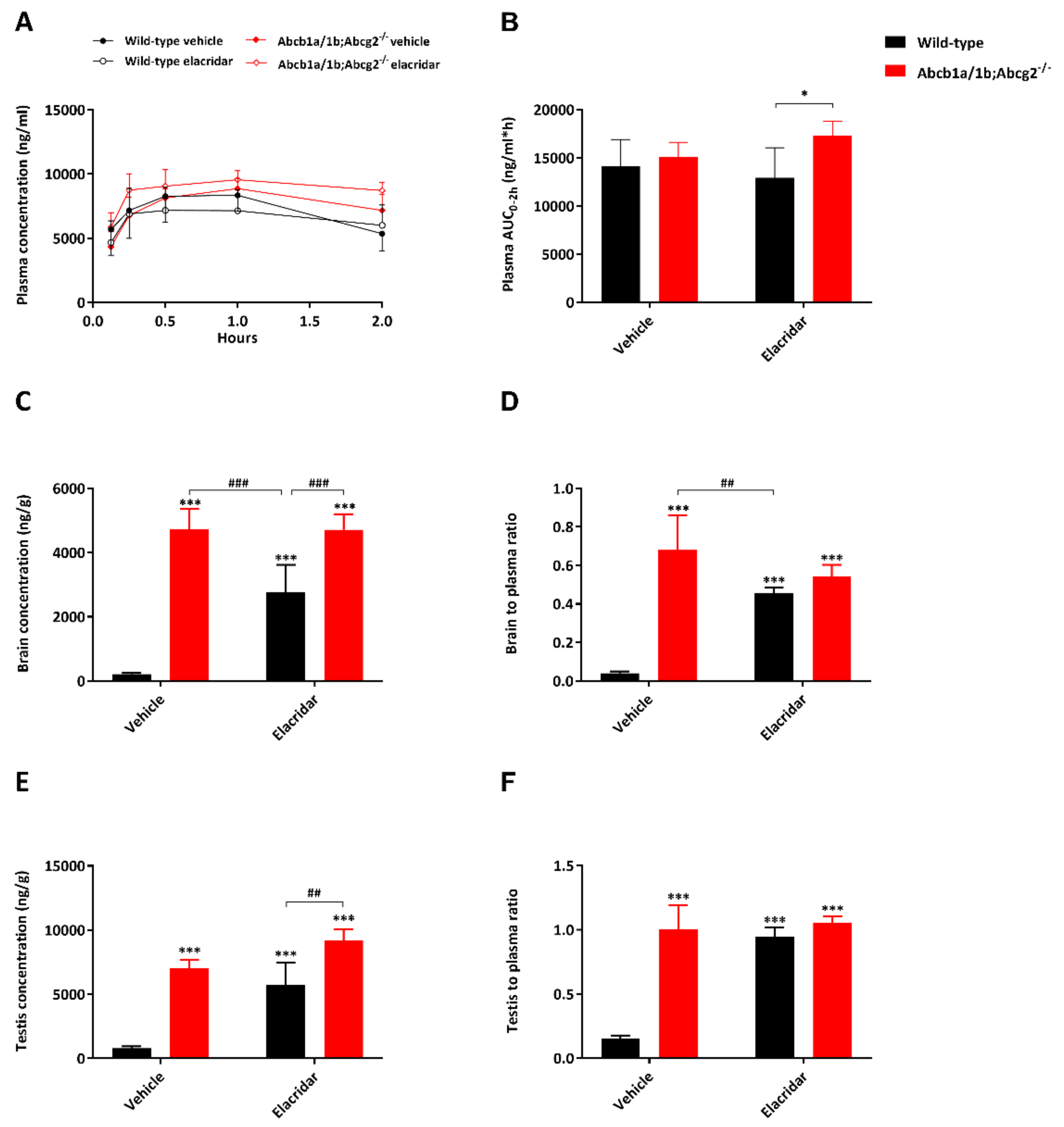
2.5. Effect of the Dual ABCB1 and ABCG2 Inhibitor Elacridar on Selpercatinib Brain Accumulation
2.6. Impact of CYP3A on Selpercatinib Plasma Exposure and Tissue Distribution
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Transport Assays
4.2. Cellular Uptake Assays
4.3. Animals
4.4. Drug Solutions
4.5. Plasma and Organ Pharmacokinetics of Selpercatinib in Ice
4.6. LC–MS/MS Analysis
4.7. Materials
4.8. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Manié, S.; Santoro, M.; Fusco, A.; Billaud, M. The RET receptor: Function in development and dysfunction in congenital malformation. Trends Genet. TIG 2001, 17, 580–589. [Google Scholar] [CrossRef]
- Ibanez, C.F. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a009134. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.H.; Oh, Y.L.; Hong, M.; Yun, J.W.; Lee, H.W.; Kim, D.; Ji, Y.; Kim, D.H.; Park, W.Y.; Shin, H.T.; et al. Identification of Driving ALK Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet. 2015, 11, e1005467. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Subbiah, V.; Marchlik, E.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin. Cancer Res. 2017, 23, 1988–1997. [Google Scholar] [CrossRef] [Green Version]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet. Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet. Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Sabari, J.K.; Siau, E.D.; Drilon, A. Targeting RET-rearranged lung cancers with multikinase inhibitors. Oncoscience 2017, 4, 23–24. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Cote, G.J. Advances in Targeting RET-Dependent Cancers. Cancer Discov. 2020, 10, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. J. Clin. Oncol. 2018, 36, 102. [Google Scholar] [CrossRef]
- Food and Drug Administration. Center for Drug Evaluation and Research of the US Department of Health and Human Service, Food and Drug Administration. Multi-Discipline Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213246s000lbl.pdf (accessed on 8 May 2020).
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2014, 14, 29–44. [Google Scholar] [CrossRef] [Green Version]
- Russel, F.G.M. Transporters: Importance in Drug Absorption, Distribution, and Removal; Springer: New York, NY, USA, 2010; pp. 27–49. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Wang, Y.; Sparidans, R.W.; Li, W.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. OATP1A/1B, CYP3A, ABCB1, and ABCG2 limit oral availability of the NTRK inhibitor larotrectinib, while ABCB1 and ABCG2 also restrict its brain accumulation. Br. J. Pharmacol. 2020, 177, 3060–3074. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein and breast cancer resistance protein restrict brigatinib brain accumulation and toxicity, and, alongside CYP3A, limit its oral availability. Pharmacol. Res. 2018, 137, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Gui, C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica 2008, 38, 778–801. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/ SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflug. Arch. Eur. J. Physiol. 2004, 447, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Meier, P.J. The superfamily of organic anion transporting polypeptides. Biochim. Et Biophys. Acta 2003, 1609, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, N.; Lockhart, A.C.; Lee, W. Role of Organic Anion-Transporting Polypeptides (OATPs) in Cancer Therapy. AAPS J. 2015, 17, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Maher, J.; Chen, C.; Klaassen, C.D. Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps). Drug Metab. Dispos. 2005, 33, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef]
- Simoff, I.; Karlgren, M.; Backlund, M.; Lindstrom, A.C.; Gaugaz, F.Z.; Matsson, P.; Artursson, P. Complete Knockout of Endogenous Mdr1 (Abcb1) in MDCK Cells by CRISPR-Cas9. J. Pharm. Sci. 2016, 105, 1017–1021. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Lin, J.J.; Filleron, T.; Ni, A.; Milia, J.; Bergagnini, I.; Hatzoglou, V.; Velcheti, V.; Offin, M.; Li, B.; et al. Frequency of Brain Metastases and Multikinase Inhibitor Outcomes in Patients With RET-Rearranged Lung Cancers. J. Thorac. Oncol. 2018, 13, 1595–1601. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Johnson, M.L.; Clifford, S.E.; Somwar, R.; Kherani, J.F.; Son, J.; Bertram, A.A.; Davare, M.A.; Gladstone, E.; Ivanova, E.V.; et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion-Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin. Cancer Res. 2020, 27, 34–42. [Google Scholar] [CrossRef]
- Tang, S.C.; Nguyen, L.N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int. J. Cancer 2014, 134, 1484–1494. [Google Scholar] [CrossRef]
- Food and Drug Administration. Center for Drug Evaluation and Research of the US Department of Health and Human Service, Food and Drug Administration. Multi-Discipline Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/202570s030lbl.pdf (accessed on 26 August 2011).
- Van Hoppe, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacol. Res. 2017, 120, 43–50. [Google Scholar] [CrossRef]
- Peters, J.; Eggers, K.; Oswald, S.; Block, W.; Lütjohann, D.; Lämmer, M.; Venner, M.; Siegmund, W. Clarithromycin is absorbed by an intestinal uptake mechanism that is sensitive to major inhibition by rifampicin: Results of a short-term drug interaction study in foals. Drug Metab. Dispos. 2012, 40, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sparidans, R.W.; Potters, S.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. ABCB1 and ABCG2, but not CYP3A4 limit oral availability and brain accumulation of the RET inhibitor pralsetinib. Pharmacol. Res. 2021, 172, 105850. [Google Scholar] [CrossRef]
- Şentürk, R.; Wang, Y.; Schinkel, A.H.; Beijnen, J.H.; Sparidans, R.W. Quantitative bioanalytical assay for the selective RET inhibitors selpercatinib and pralsetinib in mouse plasma and tissue homogenates using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2020, 1147, 122131. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Genotype | ||
|---|---|---|---|
| Wild-Type | Abcb1a/1b;Abcg2-/- | Slco1a/1b-/- | |
| AUC0–4h, ng/mL*h | 26,649 ± 6360 | 30,188 ± 7632 | 36,197 ± 5255 |
| Fold change AUC0–4h | 1.0 | 1.1 | 1.4 |
| Cmax, ng/mL | 7862 ± 1814 | 8582 ± 2160 | 11,625 ± 1614 * |
| Tmax, h | 1.8 ± 1.2 | 1.6 ± 1.2 | 1.7 ± 0.52 |
| Cbrain, ng/g | 186 ± 23 | 3454 ± 855 *** | 278 ± 42 ** |
| Fold increase Cbrain | 1.0 | 18.6 | 1.5 |
| Brain-to-plasma ratio | 0.030 ± 0.004 | 0.46 ± 0.04 *** | 0.038 ± 0.005 ** |
| Fold increase ratio | 1.0 | 15.3 | 1.3 |
| Cliver, ng/g | 17,593 ± 3471 | 19,077 ± 2696 | 19,916 ± 3174 |
| Fold increase Cliver | 1.0 | 1.1 | 1.1 |
| Liver-to-plasma ratio | 2.8 ± 0.4 | 2.6 ± 0.3 | 2.7 ± 0.4 |
| Fold increase ratio | 1.0 | 0.93 | 1.0 |
| CSIC, ng/g | 91,051 ± 22,029 | 34,929 ± 16,659 * | 140,998 ± 48,076 |
| Fold change CSIC | 1.0 | 0.38 | 1.5 |
| SIC-to-plasma ratio | 14.9 ± 4.1 | 4.5 ± 1.8 *** | 19.0 ± 5.3 |
| Fold increase ratio | 1.0 | 0.30 | 1.3 |
| Ctestis, ng/g | 730 ± 103 | 5726 ± 1535 *** | 1121 ± 107 ** |
| Fold increase Ctestis | 1.0 | 7.8 | 1.5 |
| Testis-to-plasma ratio | 0.12 ± 0.02 | 0.76 ± 0.13 *** | 0.15 ± 0.01 * |
| Fold increase ratio | 1.0 | 6.3 | 1.3 |
| Parameter | Genotype | |||
|---|---|---|---|---|
| Wild-Type | Abcb1a/1b-/- | Abcg2-/- | Abcb1a/1b;Abcg2-/- | |
| AUC0–4h, ng/mL*h | 23,670 ± 2469 | 37,024 ± 9634 ** | 39,056 ± 6710 *** | 38,986 ± 4711 *** |
| Fold change AUC0–4h | 1.0 | 1.6 | 1.7 | 1.6 |
| Cmax, ng/mL | 6908 ± 761 | 10,338 ± 2641 ** | 11,447 ± 2155 *** | 11,015 ± 1653 ** |
| Tmax, h | 1.6 ± 1.4 | 1.8 ± 1.3 | 1.4 ± 0.74 | 1.5 ± 1.3 |
| Cbrain, ng/g | 420 ± 66 | 1998 ± 233 ***### | 418 ± 102 ### | 4263 ± 853 *** |
| Fold change Cbrain | 1.0 | 4.8 | 1.0 | 10.2 |
| Brain-to-plasma ratio | 0.077 ± 0.018 | 0.23 ± 0.04 ***### | 0.052 ± 0.011 *### | 0.48 ± 0.07 *** |
| Fold change ratio | 1.0 | 3.0 | 0.68 | 6.2 |
| CLiver, ng/g | 25,737 ± 3219 | 34,014 ± 7169 ## | 19,334 ± 3483 * | 23,157 ± 3900 |
| Fold increase Cliver | 1.0 | 1.3 | 0.75 | 0.90 |
| Liver-to-plasma ratio | 4.7 ± 0.7 | 3.9 ± 1.0 # | 2.4 ± 0.3 *** | 2.6 ± 0.5 *** |
| Fold change ratio | 1.0 | 0.83 | 0.51 | 0.55 |
| CSI, ng/g | 21,649 ± 26,731 | 16,952 ± 2729 | 12,967 ± 3639 | 11,576 ± 734 |
| Fold increase CSI | 1.0 | 0.83 | 0.60 | 0.53 |
| SI-to-plasma ratio | 3.7 ± 4.3 | 2.0 ± 0.6 | 1.6 ± 0.4 | 1.3 ± 0.1 |
| Fold change ratio | 1.0 | 0.54 | 0.43 | 0.35 |
| CSIC, ng/g | 98,083 ± 38,906 | 69,396 ± 15,655 ## | 91,686 ± 35,700 ### | 27,386 ± 3533 *** |
| Fold increase CSIC | 1.0 | 0.71 | 0.93 | 0.28 |
| SIC-to-plasma ratio | 18.2 ± 8.5 | 8.1 ± 2.9 *## | 11.4 ± 3.9 ### | 3.1 ± 0.68 *** |
| Fold change ratio | 1.0 | 0.45 | 0.63 | 0.17 |
| SIC percentage of dose, % | 2.3 ± 0.9 | 1.1 ± 0.3 | 1.6 ± 0.7 # | 0.69 ± 0.52 *** |
| Fold change ratio | 1.0 | 0.48 | 0.70 | 0.30 |
| Ctestis, ng/g | 810.8 ± 148.4 | 3477 ± 634.1 ***### | 1049 ± 192.2 ### | 8328 ± 934.8 *** |
| Fold change Ctestis | 1.0 | 4.3 | 1.3 | 10.3 |
| Testis-to-plasma ratio | 0.15 ± 0.02 | 0.39 ± 0.06 ***### | 0.13 ± 0.03 ### | 0.94 ± 0.10 *** |
| Fold change ratio | 1.0 | 2.7 | 0.90 | 6.4 |
| Parameter | Genotype/Groups | |||
|---|---|---|---|---|
| Vehicle | Elacridar | |||
| Wild-Type | Abcb1a/1b;Abcg2−/− | Wild-Type | Abcb1a/1b;Abcg2−/− | |
| AUC0–2h, ng/mL*h | 14,092 ± 2816 | 15,098 ± 1503 | 12,928 ± 3121 | 17,294 ± 1513 # |
| Fold change AUC0–2h | 1.0 | 1.1 | 0.92 | 1.2 |
| Cmax, ng/mL | 8739 ± 1560 | 8865 ± 792 | 7466 ± 1848 | 9617 ± 776 # |
| Tmax, h | 0.75 ± 0.27 | 1.0 ± 0.0# | 0.50 ± 0.27 | 0.79 ± 0.33 |
| Cbrain, ng/g | 210 ± 40 | 4726 ± 638 ***### | 2765 ± 851 *** | 4713 ± 484 ***### |
| Fold increase Cbrain | 1.0 | 22.5 | 13.2 | 22.4 |
| Brain-to-plasma ratio | 0.041 ± 0.009 | 0.68 ± 0.18 ***### | 0.46 ± 0.03 *** | 0.54 ± 0.06 *** |
| Fold increase ratio | 1.0 | 17 | 11.5 | 13.5 |
| CLiver, ng/g | 18,134 ± 2604 | 21,445 ± 2330 | 20,028 ± 4435 | 23,628 ± 4827 |
| Fold increase Cliver | 1.0 | 1.2 | 1.1 | 1.3 |
| Liver-to-plasma ratio | 3.5 ± 0.8 | 3.1 ± 0.7 | 3.4 ± 0.3 | 2.7 ± 0.4 |
| Fold change ratio | 1.0 | 0.89 | 1.0 | 0.77 |
| CSI + SIC, ng/g | 37,604 ± 13,607 | 18,845 ± 3628 ** | 26,630 ± 6802 | 21,226 ± 929 * |
| Fold increase CSI + SIC | 1.0 | 0.50 | 0.71 | 0.56 |
| SI + SIC-to-plasma ratio | 7.0 ± 1.9 | 2.7 ± 0.8 ***## | 4.6 ± 1.3 * | 2.4 ± 0.2 ***## |
| Fold change ratio | 1.0 | 0.39 | 0.66 | 0.34 |
| Ctestis, ng/g | 788 ± 159 | 7020 ± 664 *** | 5709 ± 1744 *** | 9202 ± 856 ***### |
| Fold increase Ctestis | 1.0 | 8.9 | 7.2 | 11.7 |
| Testis-to-plasma ratio | 0.15 ± 0.03 | 1.0 ± 0.2 *** | 0.94 ± 0.08 *** | 1.1 ± 0.05 *** |
| Fold change ratio | 1.0 | 6.7 | 6.3 | 7.3 |
| Parameter | Genotype | ||
|---|---|---|---|
| Wild-Type | Cyp3a-/- | Cyp3aXAV | |
| AUC0–8h, ng/mL*h | 52,251 ± 6922 | 72,243 ± 5642 *** | 45,755 ± 3460 ### |
| Fold change AUC0–8h | 1.0 | 1.4 | 0.88 |
| Cmax, ng/mL | 8556 ± 1299 | 12,295 ± 1311 *** | 9477 ± 548 ## |
| Tmax, h | 1.5 ± 1.3 | 1.4 ± 1.3 | 0.29 ± 0.10 *# |
| Cbrain, ng/g | 82.4 ± 16.8 | 86.2 ± 24.2 | 51.0 ± 15.2 *## |
| Fold change Cbrain | 1.0 | 1.0 | 0.62 |
| Brain-to-plasma ratio | 0.021 ± 0.002 | 0.022 ± 0.002 | 0.022 ± 0.002 |
| Fold change ratio | 1.0 | 1.0 | 1.0 |
| CLiver, ng/g | 8255 ± 1615 | 7896 ± 1274 | 4386 ± 755 ***### |
| Fold change Cliver | 1.0 | 1.0 | 0.53 |
| Liver-to-plasma ratio | 2.1 ± 0.3 | 2.1 ± 0.2 | 2.0 ± 0.2 |
| Fold change ratio | 1.0 | 1.0 | 1.0 |
| CSI, ng/g | 7302 ± 1116 | 6384 ± 930 | 5030 ± 347 ***# |
| Fold change CSI | 1.0 | 0.87 | 0.69 |
| SI-to-plasma ratio | 1.9 ± 0.4 | 1.7 ± 0.3 | 2.3 ± 0.6 |
| Fold change ratio | 1.0 | 0.89 | 1.2 |
| CSIC, ng/g | 83,866 ± 51,955 | 44,922 ± 15,127 | 48,515 ± 13,654 |
| Fold change CSIC | 1.0 | 0.54 | 0.58 |
| SIC-to-plasma ratio | 20.0 ± 7.9 | 11.5 ± 3.3 * | 22.3 ± 7.5 ## |
| Fold change ratio | 1.0 | 0.58 | 1.1 |
| SIC percentage of dose, % | 2.5 ± 0.9 | 1.4 ± 0.6 * | 1.5 ± 0.5 |
| Fold change % | 1.0 | 0.56 | 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Sparidans, R.W.; Potters, S.; Şentürk, R.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (ABCB1/MDR1) and BCRP (ABCG2) Limit Brain Accumulation and Cytochrome P450-3A (CYP3A) Restricts Oral Exposure of the RET Inhibitor Selpercatinib (RETEVMO). Pharmaceuticals 2021, 14, 1087. https://doi.org/10.3390/ph14111087
Wang Y, Sparidans RW, Potters S, Şentürk R, Lebre MC, Beijnen JH, Schinkel AH. P-Glycoprotein (ABCB1/MDR1) and BCRP (ABCG2) Limit Brain Accumulation and Cytochrome P450-3A (CYP3A) Restricts Oral Exposure of the RET Inhibitor Selpercatinib (RETEVMO). Pharmaceuticals. 2021; 14(11):1087. https://doi.org/10.3390/ph14111087
Chicago/Turabian StyleWang, Yaogeng, Rolf W. Sparidans, Sander Potters, Rahime Şentürk, Maria C. Lebre, Jos H. Beijnen, and Alfred H. Schinkel. 2021. "P-Glycoprotein (ABCB1/MDR1) and BCRP (ABCG2) Limit Brain Accumulation and Cytochrome P450-3A (CYP3A) Restricts Oral Exposure of the RET Inhibitor Selpercatinib (RETEVMO)" Pharmaceuticals 14, no. 11: 1087. https://doi.org/10.3390/ph14111087