Psychotropic Drugs for the Management of Chronic Pain and Itch

Abstract
: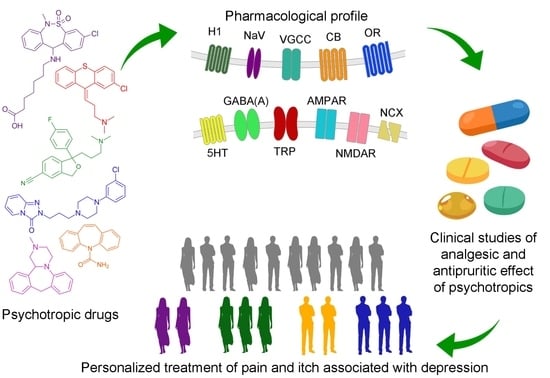
1. Overview
2. Targets of Analgesic and Antipruritic Therapy
2.1. NMDA and AMPA Receptors
2.2. Sodium Channels
2.3. Calcium Channels
2.4. GABA(A)-Receptors
2.5. Opioid Receptors
2.6. Cannabinoid Receptors
2.7. 5-HT7 Receptors
2.8. Sodium-Calcium Exchanger
2.9. Histamine Receptors
2.10. TRP Channels
3. Psychotropic Drugs for Management of Pain and Itching Syndromes and Their Interaction with the Targets for Analgesic and Antipruritic Therapy
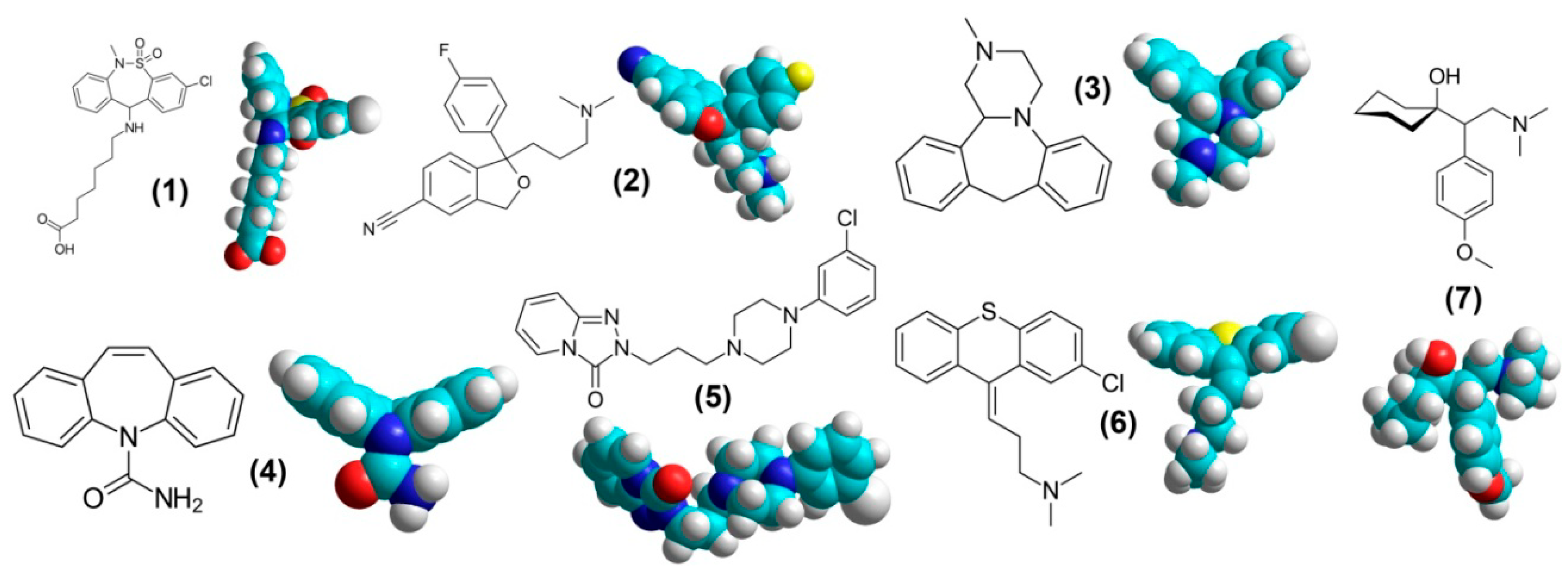
3.1. Tianeptine
3.1.1. Tianeptine. Animal Models
3.1.2. Tianeptine. Clinical Studies
3.1.3. Tianeptine. Interaction with the Receptors
3.2. Citalopram
3.2.1. Citalopram. Animal Models
3.2.2. Citalopram. Clinical Studies
3.2.3. Citalopram. Interaction with the Receptors
3.3. Mianserin
3.3.1. Mianserin. Animal Models
3.3.2. Mianserin. Clinical Studies
3.3.3. Mianserin. Interaction with the Receptors
3.4. Carbamazepine
3.4.1. Carbamazepine. Animal Models
3.4.2. Carbamazepine. Clinical Studies
3.4.3. Carbamazepine. Interaction with the Receptors
3.5. Trazodone
3.5.1. Trazodone. Animal Models
3.5.2. Trazodone. Clinical Studies
3.5.3. Trazodone. Interaction with the Receptors
3.6. Chlorprothixene
3.6.1. Chlorprothixene. Animal Models
3.6.2. Chlorprothixene. Clinical Studies
3.6.3. Chlorprothixene. Interaction with the Receptors
3.7. Venlafaxine
3.7.1. Venlafaxine. Animal Models
3.7.2. Venlafaxine. Clinical Studies
3.7.3. Venlafaxine. Interaction with the Receptors
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murtagh, F.E.; Addington-Hall, J.; Higginson, I.J. The prevalence of symptoms in end-stage renal disease: a systematic review. Adv. Chronic Kidney Dis. 2007, 14, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.K.; Nones, C.F.; Reis, R.C.; Chichorro, J.G.; Cunha, J.M. Diabetic neuropathic pain: Physiopathology and treatment. World J. Diabetes 2015, 6, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.T. Neuropathic pain in cancer. Br. J. Anaesth. 2013, 111, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, D.; Schmelz, M.; Ballantyne, J.C. Itch: Mechanisms and mediators. In Progress in Pain Research and Management; Dostrovsky, J.O., Carr, D.B., Koltzenburg, M., Eds.; IASP Press: Seattle, WA, USA, 2003; pp. 213–226. [Google Scholar]
- Carstensa, E.; Akiyama, T. Central mechanisms of itch. Curr. Probl. Dermatol. 2016, 50, 11–17. [Google Scholar]
- Ferm, I.; Sterner, M.; Wallengren, J. Somatic and psychiatric comorbidity in patients with chronic pruritus. Acta Derm. Venereol. 2010, 90, 395–400. [Google Scholar]
- Torta, R.; Ieraci, V.; Zizzi, F. A Review of the Emotional Aspects of Neuropathic Pain: From Comorbidity to Co-Pathogenesis. Pain Ther. 2017, 6, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bair, M.J.; Robinson, R.L.; Katon, W.; Kroenke, K. Depression and pain comorbidity: A literature review. Arch. Intern. Med. 2003, 163, 2433–2445. [Google Scholar] [CrossRef]
- Vanchakova, N.P.; Rybakova, K.V.; Smirnov, A.V.; Shestakova, N.N. Specific use of antidepressants of different chemical groups in patients with chronic renal failure and syndromes of itching and pain receiving chronic hemodialysis. Nephrology (Saint-Petersburg) 2003, 7, 62–65. (In Russian) [Google Scholar]
- Mika, J.; Zychowska, M.; Makuch, W.; Rojewska, E.; Przewlocka, B. Neuronal and immunological basis of action of antidepressants in chronic pain—Clinical and experimental studies. Pharmacol. Rep. 2013, 65, 1611–1621. [Google Scholar] [CrossRef]
- Yosipovitch, G.; Bernhard, J.D. Clinical practice. Chronic pruritus. N. Engl. J. Med. 2013, 368, 1625–1634. [Google Scholar] [CrossRef]
- Baltenberger, E.P.; Buterbaugh, W.M.; Martin, B.S.; Thomas, C.J. Review of antidepressants in the treatment of neuropathic pain. Ment. Health Clin. 2015, 5, 123–133. [Google Scholar] [CrossRef]
- Jensen, T.S. Anticonvulsants in neuropathic pain: Rationale and clinical evidence. Eur. J. Pain 2002, 6, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Aigner, M.; Ossege, M.; Pernicka, E.; Wildner, B.; Sycha, T. Antipsychotics for acute and chronic pain in adults. J. Pain Symptom Manag. 2010, 39, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Shestakova, N.N.; Vanchakova, N.P. Theoretical conformational analysis of antidepressant as a way for evaluation of their efficiency for pain and itch syndrome management in patients with end-stage renal disease under chronic hemodialysis. Dokl. Biochem. Biophys. 2006, 409, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Sernagor, E.; Kuhn, D.; Vyklicky, L., Jr.; Mayer, M.L. Open channel block of NMDA receptor responses evoked by tricyclic antidepressants. Neuron 1989, 2, 1221–1227. [Google Scholar] [CrossRef]
- Watanabe, Y.; Saito, H.; Abe, K. Tricyclic antidepressants block NMDA receptor-mediated synaptic responses and induction of long-term potentiation in rat hippocampal slices. Neuropharmacology 1993, 32, 479–486. [Google Scholar] [CrossRef]
- Barygin, O.I.; Gmiro, V.E.; Kim, K.K.; Magazanik, L.G.; Tikhonov, D.B. Blockade of NMDA receptor channels by 9-aminoacridine and its derivatives. Neurosci. Lett. 2009, 451, 29–33. [Google Scholar] [CrossRef]
- Barygin, O.I.; Nagaeva, E.I.; Tikhonov, D.B.; Belinskaya, D.A.; Vanchakova, N.P.; Shestakova, N.N. Inhibition of the NMDA and AMPA receptor channels by antidepressants and antipsychotics. Brain Res. 2017, 1660, 58–66. [Google Scholar] [CrossRef]
- Vanchakova, N.P.; Smirnov, A.V.; Rybakova, K.V.; Khalezova, N.B.; Shestakova, N.N. Comparative clinical studies of mianserin and carbamazepine efficacy for pain and itch management in patients with end-renal disease under chronic hemodialysis. Eur. J. Pain 2006, 10, S139. [Google Scholar] [CrossRef]
- Shestakova, N.N.; Vanchakova, N.P. The technology of detection among antidepressants and anticonvulsants the medicines for management of itch and pain syndromes using computer modeling methods. Alm. Clin. Med. 2008, 17, 256–259. (In Russian) [Google Scholar]
- Shestakova, N.N.; Belinskaya, D.A.; Barygin, O.I.; Vanchakova, N.P. The complex therapy for patients suffered from diffuse pruritus. In Proceedings of the 16th World Congress on Pain, Yokohama, Japan, 26–30 September 2016; Available online: https://event.crowdcompass.com/wcp2016/activity/B93PPmgvaU (accessed on 25 May 2019).
- Barygin, O.I.; Sechenov Institute of Evolutionary Physiology and Biochemistry, Saint Petersburg, Russia. Personal communication, 2019.
- Belinskaia, D.A.; Goncharov, N.V.; Shestakova, N.N. Mechanism of the Analgesic Action of Ppsychotropic Drugs: Interaction with the Transport Protein Albumin and NMDA-Receptors. In Proceedings of the International Conference on Receptors and Intracellular Signaling, Pushchino, Russia, 22–25 May 2017; Zinchenko, V.P., Berezhnov, A.V., Eds.; Fix-Print: Pushchino, Russia, 2017; pp. 674–679. (In Russian). [Google Scholar]
- Gillman, P.K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, M.; Salvat, E.; Muller, A.; Yalcin, I.; Barrot, M. Antidepressants and gabapentinoids in neuropathic pain: Mechanistic insights. Neuroscience 2016, 338, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Fornasari, D. Pharmacotherapy for Neuropathic Pain: A Review. Pain Ther. 2017, 6, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chincholkar, M. Analgesic mechanisms of gabapentinoids and effects in experimental pain models: A narrative review. Br. J. Anaesth. 2018, 120, 1315–1334. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.H.; McCulloch, C.E.; Steinman, M.A.; Grimes, B.A.; Johansen, K.L. Gabapentin and Pregabalin Use and Association with Adverse Outcomes among Hemodialysis Patients. J. Am. Soc. Nephrol. 2018, 29, 1970–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleakman, D.; Alt, A.; Nisenbaum, E.S. Glutamate receptors and pain. Semin. Cell Dev. Biol. 2006, 17, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, V.; Wang, R.; Ulzhöfer, B.; Luo, C.; Bardoni, R.; Bali, K.K.; Agarwal, N.; Tegeder, I.; Hildebrandt, U.; Nagy, G.G.; et al. Peripheral calcium-permeable AMPA receptors regulate chronic inflammatory pain in mice. J. Clin. Investig. 2011, 121, 1608–1623. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, I.O.; Malyshkin, A.A.; Belozertseva, I.V.; Sukhotina, I.A.; Sevostianova, N.Y.; Aliev, K.; Zvartau, E.E.; Parsons, C.G.; Danysz, W.; Bespalov, A.Y. Effects of low-affinity NMDA receptor channel blockers in two rat models of chronic pain. Neuropharmacology 2004, 47, 175–183. [Google Scholar] [CrossRef]
- Maier, C.; Dertwinkel, R.; Mansourian, N.; Hosbach, I.; Schwenkreis, P.; Senne, I.; Skipka, G.; Zenz, M.; Tegenthoff, M. Efficacy of the NMDA-receptor antagonist memantine in patients with chronic phantom limb pain—Results of a randomized double-blinded, placebo-controlled trial. Pain 2003, 103, 277–283. [Google Scholar] [CrossRef]
- Hudspith, M.J.; Harrisson, S.; Smith, G.; Bountra, C.; Elliot, P.J.; Birch, P.J.; Hunt, S.P.; Munglani, R. Effect of post-injury NMDA antagonist treatment on long-term Fos expression and hyperalgesia in a model of chronic neuropathic pain. Brain Res. 1999, 822, 220–227. [Google Scholar] [CrossRef]
- Bilsky, E.J.; Inturrisi, C.E.; Sadée, W.; Hruby, V.J.; Porreca, F. Competitive and non-competitive NMDA antagonists block the development of antinociceptive tolerance to morphine, but not to selective mu or delta opioid agonists in mice. Pain 1996, 68, 229–237. [Google Scholar] [CrossRef]
- Christoph, T.; Reissmüller, E.; Schiene, K.; Englberger, W.; Chizh, B.A. Antiallodynic effects of NMDA glycine(B) antagonists in neuropathic pain: possible peripheral mechanisms. Brain Res. 2005, 1048, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Coderre, T.J.; Van Empel, I. The utility of excitatory amino acid (EAA) antagonists as analgesic agents. I. Comparison of the antinociceptive activity of various classes of EAA antagonists in mechanical, thermal and chemical nociceptive tests. Pain 1994, 59, 345–352. [Google Scholar] [CrossRef]
- Bereiter, D.A.; Bereiter, D.F.; Hathaway, C.B. The NMDA receptor antagonist MK-801 reduces Fos-like immunoreactivity in central trigeminal neurons and blocks select endocrine and autonomic responses to corneal stimulation in the rat. Pain 1996, 64, 179–189. [Google Scholar] [CrossRef]
- Pappagallo, M. Neurological Basis of Pain; McGraw-Hill Professional: New York, NY, USA, 2005. [Google Scholar]
- Kristensen, J.D.; Svensson, B.; Gordh, T., Jr. The NMDA-receptor antagonist CPP abolishes neurogenic ‘wind-up pain’ after intrathecal administration in humans. Pain 1992, 51, 249–253. [Google Scholar] [CrossRef]
- Wallace, M.; White, A.; Grako, K.A.; Lane, R.; Cato, A.J.; Snodgrass, H.R. Randomized, double-blind, placebo-controlled, dose-escalation study: Investigation of the safety, pharmacokinetics, and antihyperalgesic activity of l-4-chlorokynurenine in healthy volunteers. Scand. J. Pain 2017, 17, 243–251. [Google Scholar] [CrossRef]
- Meymandi, M.S.; Keyhanfar, F.; Sepehri, G.R.; Heravi, G.; Yazdanpanah, O. The Contribution of NMDA Receptors in Antinociceptive Effect of Pregabalin: Comparison of Two Models of Pain Assessment. Anesth. Pain Med. 2017, 7, e14602. [Google Scholar] [CrossRef] [Green Version]
- Kato, E.; Matsuzawa, R.; Kobayashi, S.; Fukushima, T.; Maekawa, M.; Hori, Y. Effects of pregabalin on spinal d-serine content and NMDA receptor-mediated synaptic transmission in mice with neuropathic pain. Neurosci. Lett. 2017, 636, 270–275. [Google Scholar] [CrossRef]
- Pieri, C.; Recchioni, R.; Moroni, F.; Balkay, L.; Márián, T.; Trón, L.; Damjanovich, S. Ligand and voltage gated sodium channels may regulate electrogenic pump activity in human, mouse and rat lymphocytes. Biochem. Biophys. Res. Commun. 1989, 160, 999–1002. [Google Scholar] [CrossRef]
- Mantegazza, M.; Catterall, W.A. Voltage-Gated Na+ Channels: Structure, Function, and Pathophysiology. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Devor, M. Sodium channels and mechanisms of neuropathic pain. J. Pain 2006, 7, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Dick, I.E.; Brochu, R.M.; Purohit, Y.; Kaczorowski, G.J.; Martin, W.J.; Priest, B.T. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J. Pain 2007, 8, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Sheets, M.F.; Hanck, D.A. The sodium channel as a target for local anesthetic drugs. Front. Pharmacol. 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Maatuf, Y.; Geron, M.; Priel, A. The Role of Toxins in the Pursuit for Novel Analgesics. Toxins 2019, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.C.; Lewis, R.J. Structure-Function and Therapeutic Potential of Spider Venom-Derived Cysteine Knot Peptides Targeting Sodium Channels. Front. Pharmacol. 2019, 10, 366. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mis, M.A.; Estacion, M.; Dib-Hajj, S.D.; Waxman, S.G. NaV1.7 as a Pharmacogenomic Target for Pain: Moving Toward Precision Medicine. Trends Pharmacol. Sci. 2018, 39, 258–275. [Google Scholar] [CrossRef]
- Wu, Y.J.; Guernon, J.; Shi, J.; Ditta, J.; Robbins, K.J.; Rajamani, R.; Easton, A.; Newton, A.; Bourin, C.; Mosure, K.; et al. Development of New Benzenesulfonamides As Potent and Selective Nav1.7 Inhibitors for the Treatment of Pain. J. Med. Chem. 2017, 60, 2513–2525. [Google Scholar] [CrossRef]
- Kornecook, T.J.; Yin, R.; Altmann, S.; Be, X.; Berry, V.; Ilch, C.P.; Jarosh, M.; Johnson, D.; Lee, J.H.; Lehto, S.G.; et al. Pharmacologic Characterization of AMG8379, a Potent and Selective Small Molecule Sulfonamide Antagonist of the Voltage-Gated Sodium Channel NaV1.7. J. Pharmacol. Exp. Ther. 2017, 362, 146–160. [Google Scholar] [CrossRef]
- Zeng, X.; Li, P.; Chen, B.; Huang, J.; Lai, R.; Liu, J.; Rong, M. Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins 2018, 10, 64. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Mechanism of sodium channel block by local anesthetics, antiarrhythmics, and anticonvulsants. J. Gen. Physiol. 2017, 149, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Buyan, A.; Sun, D.; Corry, B. Protonation state of inhibitors determines interaction sites within voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2018, 115, E3135–E3144. [Google Scholar] [CrossRef] [Green Version]
- Striggow, F.; Ehrlich, B.E. Ligand-gated calcium channels inside and out. Curr. Opin. Cell Biol. 1996, 8, 490–495. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Davies, J.N.; Zamponi, G.W. Old proteins, developing roles: The regulation of calcium channels by synaptic proteins. Channels (Austin) 2008, 2, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Lewis, R.J.; Todorovic, S.M.; Arneric, S.P.; Snutch, T.P. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 2009, 60, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Dib-Hajj, S.; Meisler, M.H.; Pietrobon, D. Inherited neuronal ion channelopathies: New windows on complex neurological diseases. J. Neurosci. 2008, 28, 11768–11777. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Doering, C.J.; Zamponi, G.W.; Lewis, R.J. N-type calcium channel blockers: Novel therapeutics for the treatment of pain. Med. Chem. 2006, 2, 535–543. [Google Scholar] [CrossRef]
- Gohil, K.; Bell, J.R.; Ramachandran, J.; Miljanich, G.P. Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist, SNX-230 (omega-conopeptide MVIIC). Brain Res. 1994, 653, 258–266. [Google Scholar] [CrossRef]
- Catterall, W.A.; Few, A.P. Calcium channel regulation and presynaptic plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Pogrel, J.W.; Yaksh, T.L. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther. 1994, 269, 1117–1123. [Google Scholar] [PubMed]
- Matthews, E.A.; Dickenson, A.H. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain 2001, 92, 235–246. [Google Scholar] [CrossRef]
- Evans, A.R.; Nicol, G.D.; Vasko, M.R. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996, 712, 265–273. [Google Scholar] [CrossRef]
- McGivern, J.G.; McDonough, S.I. Voltage-gated calcium channels as targets for the treatment of chronic pain. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, L.; Chen, S.R.; Chen, H.; Xie, J.D.; Sirrieh, R.E.; MacLean, D.M.; Zhang, Y.; Zhou, M.H.; Jayaraman, V.; et al. The α2δ-1-NMDA Receptor Complex is Critically Involved in Neuropathic Pain Development and Gabapentin Therapeutic Actions. Cell Rep. 2018, 22, 2307–2321. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. The role of T-type calcium channels in peripheral and central pain processing. CNS Neurol. Disord. Drug Targets 2006, 5, 639–653. [Google Scholar] [CrossRef]
- Sekizawa, S.I.; French, A.S.; Torkkeli, P.H. Low-voltage-activated calcium current does not regulate the firing behavior in paired mechanosensory neurons with different adaptation properties. J. Neurophysiol. 2000, 83, 746–753. [Google Scholar] [CrossRef]
- M’Dahoma, S.; Gadotti, V.M.; Zhang, F.X.; Park, B.; Nam, J.H.; Onnis, V.; Balboni, G.; Lee, J.Y.; Zamponi, G.W. Effect of the T-type channel blocker KYS-05090S in mouse models of acute and neuropathic pain. Pflugers Arch. 2016, 468, 193–199. [Google Scholar] [CrossRef]
- Pudukulatham, Z.; Zhang, F.X.; Gadotti, V.M.; M’Dahoma, S.; Swami, P.; Tamboli, Y.; Zamponi, G.W. Synthesis and characterization of a disubstituted piperazine derivative with T-type channel blocking action and analgesic properties. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Alaoui, C.; Chemin, J.; Fechtali, T.; Lory, P. Modulation of T-type Ca2+ channels by Lavender and Rosemary extracts. PLoS ONE 2017, 12, e0186864. [Google Scholar] [CrossRef] [PubMed]
- Kerckhove, N.; Pereira, B.; Soriot-Thomas, S.; Alchaar, H.; Deleens, R.; Hieng, V.S.; Serra, E.; Lanteri-Minet, M.; Arcagni, P.; Picard, P.; et al. Efficacy and safety of a T-type calcium channel blocker in patients with neuropathic pain: A proof-of-concept, randomized, double-blind and controlled trial. Eur. J. Pain 2018, 22, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; DeLorey, T.M. GABA and glycine. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lippincott Williams & Wilkins: New York, NY, USA, 1999; pp. 335–346. [Google Scholar]
- Martin, I.L.; Dunn, S.M. GABA receptors. Tocris. Rev. 2002, 20, 1–8. [Google Scholar]
- Malan, T.P.; Mata, H.P.; Porreca, F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology 2002, 96, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Yaksh, T.L. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain 1997, 70, 15–22. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Tan, T.; Han, H.; Dong, Z. GABA(A) Receptors in the Central Nucleus of the Amygdala Are Involved in Pain- and Itch-Related Responses. J. Pain. 2016, 17, 181–189. [Google Scholar] [CrossRef]
- Knabl, J.; Witschi, R.; Hösl, K.; Reinold, H.; Zeilhofer, U.B.; Ahmadi, S.; Brockhaus, J.; Sergejeva, M.; Hess, A.; Brune, K.; et al. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 2008, 451, 330–334. [Google Scholar] [CrossRef]
- Atack, J.R.; Bayley, P.J.; Seabrook, G.R.; Wafford, K.A.; McKernan, R.M.; Dawson, G.R. L-655,708 enhances cognition in rats but is not proconvulsant at a dose selective for alpha5-containing GABAA receptors. Neuropharmacology 2006, 51, 1023–1029. [Google Scholar] [CrossRef]
- Rivas, F.M.; Stables, J.P.; Murphree, L.; Edwankar, R.V.; Edwankar, C.R.; Huang, S.; Jain, H.D.; Zhou, H.; Majumder, S.; Sankar, S.; et al. Antiseizure activity of novel gamma-aminobutyric acid (A) receptor subtype-selective benzodiazepine analogues in mice and rat models. J. Med. Chem. 2009, 52, 1795–1798. [Google Scholar] [CrossRef]
- Di Lio, A.; Benke, D.; Besson, M.; Desmeules, J.; Daali, Y.; Wang, Z.J.; Edwankar, R.; Cook, J.M.; Zeilhofer, H.U. HZ166, a novel GABAA receptor subtype-selective benzodiazepine site ligand, is antihyperalgesic in mouse models of inflammatory and neuropathic pain. Neuropharmacology 2011, 60, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Perrault, G.; Simiand, J.; Cohen, C.; Granger, P.; Depoortere, H.; Françon, D.; Avenet, P.; Schoemaker, H.; Evanno, Y.; et al. SL651498, a GABAA receptor agonist with subtype-selective efficacy, as a potential treatment for generalized anxiety disorder and muscle spasms. CNS Drug Rev. 2003, 9, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, M.; Suzuki, S.; Miura, K.; Terashima, M.; Hatsuda, S.; Sugita, S.; Murakami, H.; Nakazawa, K.; Ohara, M. A study of the effects of antidepressants on the GABAA receptor and its complex based on the drug actions on the power-spectral changes of rat hippocampal EEG induced by GABA antagonists and inverse agonists. Nihon Shinkei Seishin Yakurigaku Zasshi 1997, 17, 75–83. (In Japanese) [Google Scholar] [PubMed]
- Zheng, T.; Clarke, A.L.; Morris, M.J.; Reid, C.A.; Petrou, S.; O’Brien, T.J. Oxcarbazepine, not its active metabolite, potentiates GABAA activation and aggravates absence seizures. Epilepsia 2009, 50, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.R.; Yoon, S.Y.; Kim, H.W.; Yeo, J.H.; Kim, Y.H.; Oh, S.B. Peripheral GABAA receptor-mediated signaling facilitates persistent inflammatory hypersensitivity. Neuropharmacology 2018, 135, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Vasović, D.; Divović, B.; Treven, M.; Knutson, D.E.; Steudle, F.; Scholze, P.; Obradović, A.; Fabjan, J.; Brković, B.; Sieghart, W.; et al. Trigeminal neuropathic pain development and maintenance in rats are suppressed by a positive modulator of α6 GABAA receptors. Eur. J. Pain 2019, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- De la Luz-Cuellar, Y.E.; Rodríguez-Palma, E.J.; Franco-Enzástiga, Ú.; Salinas-Abarca, A.B.; Delgado-Lezama, R.; Granados-Soto, V. Blockade of spinal α5-GABAA receptors differentially reduces reserpine-induced fibromyalgia-type pain in female rats. Eur. J. Pharmacol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Lowney, L.I.; Pal, B.K. Stereospecific and Nonspecific Interactions of the Morphine Congener Levorphanol in Subcellular Fractions of Mouse Brain. Proc. Natl. Acad. Sci. USA 1971, 68, 1742–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janecka, A.; Fichna, J.; Janecki, T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L.; Gavériaux-Ruff, C. Exploring the opioid system by gene knockout. Prog. Neurobiol. 2002, 66, 285–306. [Google Scholar] [CrossRef]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, A.; Marsch, L.A.; Joseph, H.; Portenoy, R.K. Opioids and the treatment of chronic pain: Controversies, current status, and future directions. Exp. Clin. Psychopharmacol. 2008, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Nagar, V.R.; Birthi, P.; Salles, S.; Sloan, P.A. Opioid Use in Chronic Pain Patients with Chronic Kidney Disease: A Systematic Review. Pain Med. 2017, 18, 1416–1449. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.; Benveniste, H.; McLellan, A.T. Use and Misuse of Opioids in Chronic Pain. Annu. Rev. Med. 2018, 69, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [PubMed]
- Machelska, H.; Celik, M.Ö. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.T.; Kieffer, B.L.; Darcq, E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Yang, F.; He, S.Q.; Shechter, R.; Zhang, C.; Shu, B.; Zhang, T.; Tiwari, V.; Wang, Y.; Dong, X.; et al. Activation of Peripheral μ-opioid Receptors by Dermorphin [D-Arg2, Lys4] (1-4) Amide Leads to Modality-preferred Inhibition of Neuropathic Pain. Anesthesiology 2016, 124, 706–720. [Google Scholar] [CrossRef]
- Edwards, K.A.; Havelin, J.J.; Mcintosh, M.I.; Ciccone, H.A.; Pangilinan, K.; Imbert, I.; Largent-Milnes, T.M.; King, T.; Vanderah, T.W.; Streicher, J.M. A Kappa Opioid Receptor Agonist Blocks Bone Cancer Pain Without Altering Bone Loss, Tumor Size, or Cancer Cell Proliferation in a Mouse Model of Cancer-Induced Bone Pain. J. Pain 2018, 19, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.P.; Kong, Q.H.; Li, Y.L.; Pan, C.L.; Yu, J.; Cui, B.Q.; Wang, Y.F.; Wang, G.L.; Zhou, P.L.; Wang, L.L.; et al. The opioid receptor triple agonist DPI-125 produces analgesia with less respiratory depression and reduced abuse liability. Acta Pharmacol. Sin. 2017, 38, 977–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, J.P.; Kochan, K.E.; Nastase, A.F.; Montgomery, D.; Griggs, N.W.; Traynor, J.R.; Mosberg, H.I.; Jutkiewicz, E.M. In vivo effects of μ-opioid receptor agonist/δ-opioid receptor antagonist peptidomimetics following acute and repeated administration. Br. J. Pharmacol. 2018, 175, 2013–2027. [Google Scholar] [CrossRef]
- Minervini, V.; Lu, H.Y.; Padarti, J.; Osteicoechea, D.C.; France, C.P. Interactions between kappa and mu opioid receptor agonists: Effects of the ratio of drugs in mixtures. Psychopharmacology 2018, 235, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Spahn, V.; Del Vecchio, G.; Rodriguez-Gaztelumendi, A.; Temp, J.; Labuz, D.; Kloner, M.; Reidelbach, M.; Machelska, H.; Weber, M.; Stein, C. Opioid receptor signaling, analgesic and side effects induced by a computationally designed pH-dependent agonist. Sci. Rep. 2018, 8, 8965. [Google Scholar] [CrossRef]
- Howlett, A.C. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug. Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Ren, J.; Zhu, C.; Fu, J.; Lin, D.; Qiu, Y. Celastrol attenuates inflammatory and neuropathic pain mediated by cannabinoid receptor type 2. Int. J. Mol. Sci. 2014, 15, 13637–13648. [Google Scholar] [CrossRef]
- Toth, C.C.; Jedrzejewski, N.M.; Ellis, C.L.; Frey, W.H., II. Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol. Pain 2010, 6, 16. [Google Scholar] [CrossRef]
- Deng, L.; Cornett, B.L.; Mackie, K.; Hohmann, A.G. CB1 Knockout Mice Unveil Sustained CB2-Mediated Antiallodynic Effects of the Mixed CB1/CB2 Agonist CP55,940 in a Mouse Model of Paclitaxel-Induced Neuropathic Pain. Mol. Pharmacol. 2015, 88, 64–74. [Google Scholar] [CrossRef]
- Heimann, A.S.; Gomes, I.; Dale, C.S.; Pagano, R.L.; Gupta, A.; de Souza, L.L.; Luchessi, A.D.; Castro, L.M.; Giorgi, R.; Rioli, V.; et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 20588–20593. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.E.; Cesar-Rittenberg, P.; Hohmann, A.G. A double-blind, placebo-controlled, crossover pilot trial with extension using an oral mucosal cannabinoid extract for treatment of chemotherapy-induced neuropathic pain. J. Pain Symptom Manag. 2014, 47, 166–173. [Google Scholar] [CrossRef]
- Vanhoenacker, P.; Haegeman, G.; Leysen, J.E. 5-HT₇ receptors: Current knowledge and future prospects. Trends Pharmacol. Sci. 2000, 21, 70–77. [Google Scholar] [CrossRef]
- Hedlund, P.B.; Sutcliffe, J.G. Functional, molecular and pharmacological advances in 5-HT₇ receptor research. Trends Pharmacol. Sci. 2004, 25, 481–486. [Google Scholar] [CrossRef]
- Hedlund, P.B.; Huitron-Resendiz, S.; Henriksen, S.J.; Sutcliffe, J.G. 5-HT7 receptor inhibition and inactivation induce antidepressant like behavior and sleep pattern. Biol. Psychiatry 2005, 58, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.S.; Popova, N.K.; Lacivita, E.; Leopoldo, M.; Ponimaskin, E.G. Interplay between Serotonin 5-HT1A and 5-HT₇ Receptors in Depressive Disorders. CNS Neurosci. Ther. 2014, 20, 582–590. [Google Scholar] [CrossRef]
- Brenchat, A.; Romero, L.; García, M.; Pujol, M.; Burgueño, J.; Torrens, A.; Hamon, M.; Baeyens, J.M.; Buschmann, H.; Zamanillo, D.; et al. 5-HT7 receptor activation inhibits mechanical hypersensitivity secondary to capsaicin sensitization in mice. Pain 2009, 141, 239–247. [Google Scholar] [CrossRef]
- Brenchat, A.; Nadal, X.; Romero, L.; Ovalle, S.; Muro, A.; Sánchez-Arroyos, R.; Portillo-Salido, E.; Pujol, M.; Montero, A.; Codony, X.; et al. Pharmacological activation of 5-HT7 receptors reduces nerve injury-induced mechanical and thermal hypersensitivity. Pain 2010, 149, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Demirkaya, K.; Akgün, Ö.M.; Şenel, B.; Öncel Torun, Z.; Seyrek, M.; Lacivita, E.; Leopoldo, M.; Doğrul, A. Selective 5-HT7 receptor agonists LP 44 and LP 211 elicit an analgesic effect on formalin-induced orofacial pain in mice. J. Appl. Oral Sci. 2016, 24, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Santello, M.; Bisco, A.; Nevian, N.E.; Lacivita, E.; Leopoldo, M.; Nevian, T. The brain-penetrant 5-HT7 receptor agonist LP-211 reduces the sensory and affective components of neuropathic pain. Neurobiol. Dis. 2017, 106, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.; Hilton, E.; Ahmed, T.; Tidgewell, K.; Kolber, B. Understanding the role of serotonin receptor subtype 7 (5-HT7) in comorbid pain and depression using novel compounds derived from marine cyanobacteria. J. Pain 2017, 18, S17. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of Brain Na/Ca-Exchanger: From Molecular Biology to Therapeutic Perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, P.A.; Beauchamp, G.; Elie, R. Tricyclic antidepressants inhibit voltage-dependent calcium channels and Na(+)-Ca2+ exchange in rat brain cortex synaptosomes. Can. J. Physiol. Pharmacol. 1990, 68, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Watanabe, Y.; Kita, S.; Blaustein, M.P. Na+/Ca2+ exchange inhibitors: A new class of calcium regulators. Cardiovasc. Hematol. Disord. Drug Targets 2007, 7, 188–198. [Google Scholar] [CrossRef]
- Datta, S.; Waghray, T.; Torres, M.; Glusman, S. Amiodarone decreases heat, cold, and mechanical hyperalgesia in a rat model of neuropathic pain. Anesth. Analg. 2004, 98, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Singh, N. Therapeutic targets for the management of peripheral nerve injury-induced neuropathic pain. CNS Neurol. Disord. Drug Targets 2011, 10, 589–609. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Abushik, P.A.; Poguzhelskaya, E.E.; Bolshakov, K.V.; Antonov, S.M. Inhibition of plasma membrane Na/Ca-exchanger by KB-R7943 or lithium reveals its role in Ca-dependent N-methyl-D-aspartate receptor inactivation. J. Pharmacol. Exp. Ther. 2015, 355, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Poguzhelskaya, E.E.; Antonov, S.M. Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: A role of membrane cholesterol. BMC Neurosci. 2018, 19, 73. [Google Scholar] [CrossRef]
- Liu, T.; Ji, R.R. New insights into the mechanisms of itch: Are pain and itch controlled by distinct mechanisms? Pflugers Arch. 2013, 465, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Han, J.S.; Lee, K.; Bang, J.; Lee, H. The peripheral and central mechanisms underlying itch. BMB Rep. 2016, 49, 474–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; Schunack, W.; Levi, R.; Haas, H.L. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 1997, 49, 253–278. [Google Scholar] [PubMed]
- Parsons, M.E.; Ganellin, C.R. Histamine and its receptors. Br. J. Pharmacol. 2006, 147, S127–S135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossbach, K.; Nassenstein, C.; Gschwandtner, M.; Schnell, D.; Sander, K.; Seifert, R.; Stark, H.; Kietzmann, M.; Bäumer, W. Histamine H1, H3 and H4 receptors are involved in pruritus. Neuroscience 2011, 190, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Hirasawa, N. The role of histamine H1 and H4 receptors in atopic dermatitis: From basic research to clinical study. Allergol. Int. 2014, 63, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Dhand, A.; Aminoff, M.J. The neurology of itch. Brain 2014, 137, 313–322. [Google Scholar] [CrossRef]
- O’Donoghue, M.; Tharp, M.D. Antihistamines and their role as antipruritics. Dermatol Ther. 2005, 18, 333–340. [Google Scholar] [CrossRef]
- Borowy, C.S.; Mukherji, P. Antihistamine Toxicity; StatPearls Publishing: Treasure Island, FL, USA, 2018; NBK482318. [Google Scholar]
- Procopiou, P.A.; Ford, A.J.; Gore, P.M.; Looker, B.E.; Hodgson, S.T.; Holmes, D.S.; Vile, S.; Clark, K.L.; Saunders, K.A.; Slack, R.J.; et al. Design of Phthalazinone Amide Histamine H1 Receptor Antagonists for Use in Rhinitis. ACS Med. Chem. Lett. 2017, 8, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Ledneczki, I.; Tapolcsányi, P.; Gábor, E.; Éles, J.; Greiner, I.; Schmidt, É.; Némethy, Z.; Kedves, R.S.; Balázs, O.; Román, V.; et al. Discovery of novel steroidal histamine H3 receptor antagonists/inverse agonists. Bioorg. Med. Chem. Lett. 2017, 27, 4525–4530. [Google Scholar] [CrossRef]
- Ko, K.; Kim, H.J.; Ho, P.S.; Lee, S.O.; Lee, J.E.; Min, C.R.; Kim, Y.C.; Yoon, J.H.; Park, E.J.; Kwon, Y.J.; et al. Discovery of a Novel Highly Selective Histamine H4 Receptor Antagonist for the Treatment of Atopic Dermatitis. J. Med. Chem. 2018, 61, 2949–2961. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, Y.; Yang, J. Structural biology of TRP channels. Adv. Exp. Med. Biol. 2011, 704, 1–23. [Google Scholar] [PubMed]
- Basso, L.; Altier, C. Transient Receptor Potential Channels in neuropathic pain. Curr. Opin. Pharmacol. 2017, 32, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; Szallasi, A. Targeting nociceptive transient receptor potential channels to treat chronic pain: Current state of the field. Br. J. Pharmacol. 2018, 175, 2185–2203. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Hu, H. TRP Channels as Drug Targets to Relieve Itch. Pharmaceuticals 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Kittaka, H.; Yamanoi, Y.; Tominaga, M. Transient receptor potential vanilloid 4 (TRPV4) channel as a target of crotamiton and its bimodal effects. Pflugers Arch. 2017, 469, 1313–1323. [Google Scholar] [CrossRef]
- Akiyama, T.; Ivanov, M.; Nagamine, M.; Davoodi, A.; Carstens, M.I.; Ikoma, A.; Cevikbas, F.; Kempkes, C.; Buddenkotte, J.; Steinhoff, M.; et al. Involvement of TRPV4 in Serotonin-Evoked Scratching. J. Investig. Dermatol. 2016, 136, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Fang, Q.; Wang, Z.; Zhang, J.Y.; MacLeod, A.S.; Hall, R.P.; Liedtke, W.B. Transient Receptor Potential Vanilloid 4 Ion Channel Functions as a Pruriceptor in Epidermal Keratinocytes to Evoke Histaminergic Itch. J. Biol. Chem. 2016, 291, 10252–10262. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Feng, J.; Yu, G.; Yang, P.; Mack, M.R.; Du, J.; Yu, W.; Qian, A.; Zhang, Y.; Liu, S.; et al. Transient receptor potential vanilloid 4-expressing macrophages and keratinocytes contribute differentially to allergic and nonallergic chronic itch. J. Allergy Clin. Immunol. 2018, 141, 608–619. [Google Scholar] [CrossRef]
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338, 1–18. [Google Scholar] [CrossRef]
- Alves, L.A.; Bezerra, R.J.; Faria, R.X.; Ferreira, L.G.; da Silva Frutuoso, V. Physiological roles and potential therapeutic applications of the P2 × 7 receptor in inflammation and pain. Molecules 2013, 18, 10953–10972. [Google Scholar] [CrossRef] [PubMed]
- Obata, H. Analgesic Mechanisms of Antidepressants for Neuropathic Pain. Int. J. Mol. Sci. 2017, 18, 2483. [Google Scholar] [CrossRef]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; López García, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157, S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Casals-Diaz, L.; Zurawski, T.; Meng, J.; Moriarty, O.; Nealon, J.; Edupuganti, O.P.; Dolly, O. A novel therapeutic with two SNAP-25 inactivating proteases shows long-lasting anti-hyperalgesic activity in a rat model of neuropathic pain. Neuropharmacology 2017, 118, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.M.; Lee, S.H.; Jeong, H.J.; Lee, H.G.; Choi, J.I.; Yoon, M.H. The analgesic activity of intrathecal tianeptine, an atypical antidepressant, in a rat model of inflammatory pain. Anesth. Analg. 2012, 114, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Heo, B.H.; Shin, J.Y.; Park, K.S.; Lee, H.G.; Choi, J.I.; Yoon, M.H.; Kim, W.M. Effects of tianeptine on the development and maintenance of mechanical allodynia in a rat model of neuropathic pain. Neurosci. Lett. 2016, 633, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Kim, Y.H.; Jo, H.U.; Kwak, J.A.; Park, H.J. Tianeptine Reduces Mechanical Allodynia in Spinal Nerve-ligated and Chemotherapy-induced Neuropathic Mice. Pain Physician 2017, 20, E593–E600. [Google Scholar] [PubMed]
- Lee, H.; Im, J.; Won, H.; Nam, W.; Kim, Y.O.; Lee, S.W.; Lee, S.; Cho, I.H.; Kim, H.K.; Kwon, J.T.; et al. Effects of tianeptine on symptoms of fibromyalgia via BDNF signaling in a fibromyalgia animal model. Korean J. Physiol. Pharmacol. 2017, 21, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Zahran, T.; Schier, J.; Glidden, E.; Kieszak, S.; Law, R.; Bottei, E.; Aaron, C.; King, A.; Chang, A. Characteristics of Tianeptine Exposures Reported to the National Poison Data System—United States, 2000–2017. MMWR 2018, 67, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Bateup, H.; Qi, H.; Takamiya, K.; Huganir, R.L.; Spedding, M.; Roth, B.L.; McEwen, B.S.; Greengard, P. Involvement of AMPA receptor phosphorylation in antidepressant actions with special reference to tianeptine. Eur. J. Neurosci. 2007, 26, 3509–3517. [Google Scholar] [CrossRef] [PubMed]
- Gassaway, M.M.; Rives, M.L.; Kruegel, A.C.; Javitch, J.A.; Sames, D. The atypical antidepressant and neurorestorative agent tianeptine is a μ-opioid receptor agonist. Transl. Psychiatry 2014, 4, e411. [Google Scholar] [CrossRef] [PubMed]
- Bilge, S.S.; İlkaya, F.; Darakcı, Ö.; Çiftcioğlu, E.; Bozkurt, A. Opioid Receptors Contribute to Antinociceptive Effect of Tianeptine on Colorectal Distension-Induced Visceral Pain in Rats. Pharmacology 2018, 101, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Heo, B.H.; Kim, W.M.; Kim, Y.C.; Yoon, M.H. Antiallodynic effect of tianeptine via modulation of the 5-HT7 receptor of GABAergic interneurons in the spinal cord of neuropathic rats. Neurosci. Lett. 2015, 598, 91–95. [Google Scholar] [CrossRef]
- Kato, G.; Weitsch, A.F. Neurochemical profile of tianeptine, a new antidepressant drug. Clin. Neuropharmacol. 1988, 11, S43–S50. [Google Scholar] [PubMed]
- Ardid, D.; Marty, H.; Fialip, J.; Privat, A.M.; Eschalier, A.; Lavarenne, J. Comparative effects of different uptake inhibitor antidepressants in two pain tests in mice. Fundam. Clin. Pharmacol. 1992, 6, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Bomholt, S.F.; Mikkelsen, J.D.; Blackburn-Munro, G. Antinociceptive effects of the antidepressants amitriptyline, duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology 2005, 48, 252–263. [Google Scholar] [CrossRef]
- Sindrup, S.H.; Bjerre, U.; Dejgaard, A.; Brøsen, K.; Aaes-Jørgensen, T.; Gram, L.F. The selective serotonin reuptake inhibitor citalopram relieves the symptoms of diabetic neuropathy. Clin. Pharmacol. Ther. 1992, 52, 547–552. [Google Scholar] [CrossRef]
- Nørregaard, J.; Volkmann, H.; Danneskiold-Samsøe, B. A randomized controlled trial of citalopram in the treatment of fibromyalgia. Pain 1995, 61, 445–449. [Google Scholar] [CrossRef]
- Bendtsen, L.; Jensen, R.; Olesen, J. A non-selective (amitriptyline), but not a selective (citalopram), serotonin reuptake inhibitor is effective in the prophylactic treatment of chronic tension-type headache. J. Neurol. Neurosurg. Psychiatry 1996, 61, 285–290. [Google Scholar] [CrossRef]
- Anderberg, U.M.; Marteinsdottir, I.; von Knorring, L. Citalopram in patients with fibromyalgia—A randomized, double-blind, placebo-controlled study. Eur. J. Pain 2000, 4, 27–35. [Google Scholar] [CrossRef]
- Aragona, M.; Bancheri, L.; Perinelli, D.; Tarsitani, L.; Pizzimenti, A.; Conte, A.; Inghilleri, M. Randomized double-blind comparison of serotonergic (Citalopram) versus noradrenergic (Reboxetine) reuptake inhibitors in outpatients with somatoform, DSM-IV-TR pain disorder. Eur. J. Pain 2005, 9, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Kosmidou, M.; Sarmas, I.; Markoula, S.; Pelidou, S.H.; Lagos, G.; Kyritsis, A.P. Patient compliance with SSRIs and gabapentin in painful diabetic neuropathy. Clin. J. Pain 2007, 23, 267–269. [Google Scholar] [CrossRef] [PubMed]
- D’Erme, A.M.; Zanieri, F.; Campolmi, E.; Santosuosso, U.; Betti, S.; Agnoletti, A.F.; Cossidente, A.; Lotti, T. Therapeutic implications of adding the psychotropic drug escitalopram in the treatment of patients suffering from moderate-severe psoriasis and psychiatric comorbidity: A retrospective study. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Thériault, O.; Poulin, H.; Beaulieu, J.M.; Chahine, M. Differential modulation of Nav1.7 and Nav1.8 channels by antidepressant drugs. Eur. J. Pharmacol. 2015, 764, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Witchel, H.J.; Pabbathi, V.K.; Hofmann, G.; Paul, A.A.; Hancox, J.C. Inhibitory actions of the selective serotonin re-uptake inhibitor citalopram on HERG and ventricular L-type calcium currents. FEBS Lett. 2002, 512, 59–66. [Google Scholar] [CrossRef]
- Hamplová-Peichlová, J.; Krůsek, J.; Paclt, I.; Slavícek, J.; Lisá, V.; Vyskocil, F. Citalopram Inhibits L-type Calcium Channel Current in Rat Cardiomyocytes in Culture. Physiol. Res. 2002, 51, 317–321. [Google Scholar] [PubMed]
- Zahradník, I.; Minarovic, I.; Zahradníková, A. Inhibition of the cardiac L-type calcium channel current by antidepressant drugs. J. Pharmacol. Exp. Ther. 2008, 324, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Hyttel, J. Citalopram—Pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 1982, 6, 277–295. [Google Scholar] [CrossRef]
- Hesketh, S.A.; Brennan, A.K.; Jessop, D.S.; Finn, D.P. Effects of chronic treatment with citalopram on cannabinoid and opioid receptor-mediated G-protein coupling in discrete rat brain regions. Psychopharmacology 2008, 198, 29–36. [Google Scholar] [CrossRef]
- Owens, M.J.; Knight, D.L.; Nemeroff, C.B. Second-generation SSRIs: Human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol. Psychiatry 2001, 50, 345–350. [Google Scholar] [CrossRef]
- Zarrindast, M.R.; Alaei-Nia, K.; Shafizadeh, M. On the mechanism of tolerance to morphine-induced Straub tail reaction in mice. Pharmacol. Biochem. Behav. 2001, 69, 419–424. [Google Scholar] [CrossRef]
- Pakulska, W.; Czarnecka, E. Influence of mianserin on the antinociceptive effect of morphine, metamizol and indomethacin in mice. Pharmacol. Res. 2002, 46, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Üçel, U.İ.; Can, Ö.D.; Demir Özkay, Ü.; Öztürk, Y. Antihyperalgesic and antiallodynic effects of mianserin on diabetic neuropathic pain: A study on mechanism of action. Eur. J. Pharmacol. 2015, 756, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.H.G.; Broekkamp, C.L.E. A peripheral 5-HT1D-like receptor involved in serotonergic induced hindlimb scratching in rats. Eur. J. Pharmacol. 1991, 94, 201–208. [Google Scholar] [CrossRef]
- Langemark, M.; Loldrup, D.; Bech, P.; Olesen, J. Clomipramine and mianserin in the treatment of chronic tension headache. A double-blind, controlled study. Headache 1990, 30, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Van Houdenhove, B.; Verstraeten, D.; Onghena, P.; De Cuyper, H. Chronic idiopathic pain, mianserin and ‘masked’ depression. Psychother. Psychosom. 1992, 58, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Onghena, P.; De Cuyper, H.; Van Houdenhove, B.; Verstraeten, D. Mianserin and chronic pain: A double-blind placebo-controlled process and outcome study. Acta Psychiatr. Scand. 1993, 88, 198–204. [Google Scholar] [CrossRef]
- Manna, V.; Bolino, F.; Di Cicco, L. Chronic tension-type headache, mood depression and serotonin: Therapeutic effects of fluvoxamine and mianserin. Headache 1994, 34, 44–49. [Google Scholar] [CrossRef]
- Tohda, M.; Urushihara, H.; Nomura, Y. Inhibitory effects of antidepressants on NMDA-induced currents in Xenopus oocytes injected with rat brain RNA. Neurochem. Int. 1995, 26, 53–58. [Google Scholar] [CrossRef]
- Lenkey, N.; Karoly, R.; Lukacs, P.; Vizi, E.S.; Sunesen, M.; Fodor, L.; Mike, A. Classification of drugs based on properties of sodium channel inhibition: A comparative automated patch-clamp study. PLoS ONE 2010, 5, e15568. [Google Scholar] [CrossRef]
- Lazar, A.; Lenkey, N.; Pesti, K.; Fodor, L.; Mike, A. Different pH-sensitivity patterns of 30 sodium channel inhibitors suggest chemically different pools along the access pathway. Front. Pharmacol. 2015, 6, 210. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Morel, N.; Vanbellinghen, A.M.; Lebrun, P. Blockade of calcium entry in smooth muscle cells by the antidepressant imipramine. Biochem. Pharmacol. 2004, 68, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Boselli, C.; Santagostino-Barbone, M.; Lucchelli, A. Older versus newer antidepressants: Substance P or calcium antagonism? Can. J. Physiol. Pharmacol. 2007, 85, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Blier, P.; de Montigny, C.; Tardif, D. Effects of the two antidepressant drugs mianserin and indalpine on the serotonergic system: Single-cell studies in the rat. Psychopharmacology 1984, 84, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Olianas, M.C.; Dedoni, S.; Onali, P. The atypical antidepressant mianserin exhibits agonist activity at κ-opioid receptors. Br. J. Pharmacol. 2012, 167, 1329–1341. [Google Scholar] [CrossRef]
- Schreiber, S.; Backer, M.M.; Kaufman, J.P.; Pick, C.G. Interaction between the tetracyclic antidepressant mianserin HCl and opioid receptors. Eur. Neuropsychopharmacol. 1998, 8, 297–302. [Google Scholar] [CrossRef]
- Lucchelli, A.; Santagostino-Barbone, M.G.; D’Agostino, G.; Masoero, E.; Tonini, M. The interaction of antidepressant drugs with enteric 5-HT7 receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 284–289. [Google Scholar] [CrossRef]
- Röser, C.; Jordan, N.; Balfanz, S.; Baumann, A.; Walz, B.; Baumann, O.; Blenau, W. Pharmacological Characterization of Serotonin 5-HT2α and 5-HT7 Receptors in the Salivary Glands of the Blowfly Calliphora vicina. PLoS ONE 2012, 7, e49459. [Google Scholar] [CrossRef]
- Hall, H.; Ogren, S.O. Effects of antidepressant drugs on histamine-H1 receptors in the brain. Life Sci. 1984, 34, 597–605. [Google Scholar] [CrossRef]
- Kanba, S.; Richelson, E. Histamine H1 receptors in human brain labelled with [3H]doxepin. Brain Res. 1984, 304, 1–7. [Google Scholar] [CrossRef]
- Nguyen, T.; Shapiro, D.A.; George, S.R.; Setola, V.; Lee, D.K.; Cheng, R.; Rauser, L.; Lee, S.P.; Lynch, K.R.; Roth, B.L.; et al. Discovery of a novel member of the histamine receptor family. Mol. Pharmacol. 2001, 59, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Rossoni, G.; Sacerdote, P.; Panerai, A.E.; Berti, F. Carbamazepine exerts anti-inflammatory effects in the rat. Eur. J. Pharmacol. 1995, 294, 71–74. [Google Scholar] [CrossRef]
- Sakaue, A.; Honda, M.; Tanabe, M.; Ono, H. Antinociceptive effects of sodium channel-blocking agents on acute pain in mice. J. Pharmacol. Sci. 2004, 95, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Tsuji, M.; Takeda, H.; Harada, Y.; Nohara, J.; Matsumiya, T.; Chiba, H. Antidepressants enhance the antinociceptive effects of carbamazepine in the acetic acid-induced writhing test in mice. J. Pharmacol. 2006, 550, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Chogtu, B.; Bairy, K.L.; Smitha, D.; Dhar, S.; Himabindu, P. Comparison of the efficacy of carbamazepine, gabapentin and lamotrigine for neuropathic pain in rats. Indian J. Pharmacol. 2011, 43, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Mohajjel Nayebi, A.; Sharifi, H.; Ramadzani, M.; Rezazadeh, H. Effect of acute and chronic administration of carbamazepine on Cisplatin-induced hyperalgesia in rats. Jundishapur J. Nat. Pharm. Prod. 2012, 7, 27–30. [Google Scholar] [CrossRef]
- Hama, A.T.; Pearson, J.P.; Sagen, J. Effects of repeated dosing with mechanistically distinct antinociceptive ligands in a rat model of neuropathic spinal cord injury pain. Pharmacol. Res. Perspect. 2014, 2, e00034. [Google Scholar] [CrossRef]
- Deseure, K.; Hans, G.H. Differential drug effects on spontaneous and evoked pain behavior in a model of trigeminal neuropathic pain. J. Pain Res. 2017, 28, 279–286. [Google Scholar] [CrossRef]
- Campbell, F.G.; Graham, J.G.; Zilkha, K.J. Clinical trial of carbazepine (tegretol) in trigeminal neuralgia. J. Neurol. Neurosurg. Psychiatry 1966, 29, 265–267. [Google Scholar] [CrossRef]
- Taylor, J.C.; Brauer, S.; Espir, M.L. Long-term treatment of trigeminal neuralgia with carbamazepine. Postgrad. Med. J. 1981, 57, 16–18. [Google Scholar] [CrossRef]
- Qi, L.; Liu, C.; Huang, M. A double-blind study of the effect of phenytoin on trigeminal neuralgia. Presented at the National Workshop of Clinical Use of Phenytoin, Chengdu, China, 1995, 3225. Available online: http://www.remarkablemedicine.com/Clinical/clinicaluses/paintreatment/trigeminal.html (accessed on 25 May 2019).
- Wilton, T.D. Tegretol in the treatment of diabetic neuropathy. S. Afr. Med. J. 1974, 48, 869–872. [Google Scholar] [PubMed]
- Saeed, T.; Nasrullah, M.; Ghafoor, A.; Shahid, R.; Islam, N.; Khattak, M.U.; Maheshwary, N.; Siddiqi, A.; Khan, M.A. Efficacy and tolerability of carbamazepine for the treatment of painful diabetic neuropathy in adults: A 12-week, open-label, multicenter study. Int. J. Gen. Med. 2014, 7, 339–343. [Google Scholar] [PubMed]
- Yamamoto, M.; Yabuki, S.; Hayabara, T.; Otsuki, S. Paroxysmal itching in multiple sclerosis: A report of three cases. J. Neurol. Neurosurg. Psychiatry 1981, 44, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Tait, C.P.; Grigg, E.; Quirk, C.J. Brachioradial pruritus and cervical spine manipulation. Australas. J. Dermatol. 1998, 39, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, M.; Parry, E.J.; Telfer, N.R. Trigeminal trophic syndrome: Successful treatment with carbamazepine. Br. J. Dermatol. 1999, 141, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Basselin, M.; Villacreses, N.E.; Chen, M.; Bell, J.M.; Rapoport, S.I. Chronic carbamazepine administration reduces N-methyl-D-aspartate receptor-initiated signaling via arachidonic acid in rat brain. Biol. Psychiatry 2007, 62, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Rezvanfard, M.; Zarrindast, M.R.; Bina, P. Role of ventral hippocampal GABA(A) and NMDA receptors in the anxiolytic effect of carbamazepine in rats using the elevated plus maze test. Pharmacology 2009, 84, 356–366. [Google Scholar] [CrossRef]
- Matsumoto, A.; Arisaka, H.; Hosokawa, Y.; Sakuraba, S.; Sugita, T.; Umezawa, N.; Kaku, Y.; Yoshida, K.; Kuwana, S. Effect of carbamazepine and gabapentin on excitability in the trigeminal subnucleus caudalis of neonatal rats using a voltage-sensitive dye imaging technique. Biol. Res. 2015, 48, 36. [Google Scholar] [CrossRef]
- Willow, M.; Gonoi, T.; Catterall, W.A. Voltage-clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in neuroblastoma cells. Mol. Pharmacol. 1985, 27, 549–558. [Google Scholar]
- McLean, M.J.; Macdonald, R.L. Carbamazepine and 10,11-epoxycarbamazepine produce use- and voltage-dependent limitation of rapidly firing action potentials of mouse central neurons in cell culture. J. Pharmacol. Exp. Ther. 1986, 238, 727–738. [Google Scholar]
- Ragsdale, S.; Scheuer, T.; Catterall, W.A. Frequency and voltage-dependent inhibition of type IIA Na channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic and anticonvulsant drugs. Mol. Pharmacol. 1994, 40, 756–765. [Google Scholar]
- Kuo, C.C.; Chen, R.S.; Lu, L.; Chen, R.C. Carbamazepine inhibition of neuronal Na currents: Quantitative distinction from phenytoin and possible therapeutic implications. Mol. Pharmacol. 1997, 51, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Bean, B.P. Sidedness of carbamazepine accessibility to voltage-gated sodium channels. Mol. Pharmacol. 2014, 85, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Walden, J.; Grunze, H.; Bingmann, D.; Liu, Z.; Düsing, R. Calcium antagonistic effects of carbamazepine as a mechanism of action in neuropsychiatric disorders: Studies in calcium dependent model epilepsies. Eur. Neuropsychopharmacol. 1992, 2, 455–462. [Google Scholar] [CrossRef]
- Schumacher, T.B.; Beck, H.; Steinhäuser, C.; Schramm, J.; Elger, C.E. Effects of phenytoin, carbamazepine, and gabapentin on calcium channels in hippocampal granule cells from patients with temporal lobe epilepsy. Epilepsia 1998, 39, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Granger, P.; Biton, B.; Faure, C.; Vige, X.; Depoortere, H.; Graham, D.; Langer, S.Z.; Scatton, B.; Avenet, P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995, 47, 1189–1196. [Google Scholar] [PubMed]
- Jackson, H.C.; Nutt, D.J. Investigation of the involvement of opioid receptors in the action of anticonvulsants. Psychopharmacology 1993, 111, 486–490. [Google Scholar] [CrossRef]
- Due, M.R.; Yang, X.-F.; Allette, Y.M.; Randolph, A.L.; Ripsch, M.S.; Wilson, S.M.; Dustrude, E.T.; Khanna, R.; White, F.A. Carbamazepine Potentiates the Effectiveness of Morphine in a Rodent Model of Neuropathic Pain. PLoS ONE 2014, 9, e107399. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Gallelli, L.; Avenoso, T.; Squillace, A.; De Sarro, G. Effects of carbamazepine/oxycodone coadministration in the treatment of trigeminal neuralgia. Ann. Pharmacother. 2011, 45, e33. [Google Scholar] [CrossRef]
- Tomić, M.A.; Pecikoza, U.B.; Micov, A.M.; Stepanović-Petrović, R.M. The Efficacy of Eslicarbazepine Acetate in Models of Trigeminal, Neuropathic, and Visceral Pain: The Involvement of 5-HT1B/1D Serotonergic and CB1/CB2 Cannabinoid Receptors. Anesth. Analg. 2015, 121, 1632–1639. [Google Scholar] [CrossRef]
- Oláh, Z.; Jósvay, K.; Pecze, L.; Letoha, T.; Babai, N.; Budai, D.; Otvös, F.; Szalma, S.; Vizler, C. Anti-calmodulins and tricyclic adjuvants in pain therapy block the TRPV1 channel. PLoS ONE 2007, 2, e545. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Backer, M.M.; Herman, I.; Shamir, D.; Boniel, T.; Pick, C.G. The antinociceptive effect of trazodone in mice is mediated through both µ-opioid and serotonergic mechanisms. Behav. Brain Res. 2000, 114, 51–56. [Google Scholar] [CrossRef]
- Okuda, K.; Takanishi, T.; Yoshimoto, K.; Ueda, S. Trazodone hydrochloride attenuates thermal hyperalgesia in achronic constriction injury rat model. Eur. J. Anaesthesiol. 2003, 20, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Nagata, T.; Hayashi, T.; Miyata, M.; Kawakami, Y. Intracerebroventricular injection of trazodone produces 5-HT receptor subtype mediated anti-nociception at the supraspinal and spinal levels. Eur. Neuropsychopharmacol. 2004, 14, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C. The use of low-dose trazodone in the treatment of painful diabetic neuropathy. J. Am. Podiatr. Med. Assoc. 1999, 89, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Morillas-Arques, P.; Rodriguez-Lopez, C.M.; Molina-Barea, R.; Rico-Villademoros, F.; Calandre, E.P. Trazodone for the treatment of fibromyalgia: An open-label, 12-week study. BMC Musculoskelet. Disord. 2010, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Calandre, E.P.; Morillas-Arques, P.; Molina-Barea, R.; Rodriguez-Lopez, C.M.; Rico-Villademoros, F. Trazodone plus pregabalin combination in the treatment of fibromyalgia: A two-phase, 24-week, open-label uncontrolled study. BMC Musculoskelet. Disord. 2011, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Pancrazio, J.J.; Kamatchi, G.L.; Roscoe, A.K.; Lynch, C., III. Inhibition of neuronal Na+ channels by antidepressant drugs. J. Pharmacol. Exp. Ther. 1998, 284, 208–214. [Google Scholar] [PubMed]
- Lee, S.; Lee, H.A.; Kim, S.J.; Kim, K.S. Cellular mechanisms for trazodone-induced cardiotoxicity. Hum. Exp. Toxicol. 2016, 35, 501–510. [Google Scholar] [CrossRef]
- Kraus, R.L.; Li, Y.; Jovanovska, A.; Renger, J.J. Trazodone inhibits T-type calcium channels. Neuropharmacology 2007, 53, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Richelson, E.; Nelson, A. Antagonism by antidepressants of neurotransmitter receptors of normal human brain in vitro. J. Pharmacol. Exp. Ther. 1984, 230, 94–102. [Google Scholar] [PubMed]
- Scholl, B.; Burge, J.; Priebe, N.J. Binocular integration and disparity selectivity in mouse primary visual cortex. J. Neurophysiol. 2013, 109, 3013–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantirigama, M.L.; Huang, H.H.; Bekkers, J.M. Spontaneous activity in the piriform cortex extends the dynamic range of cortical odor coding. Proc. Natl. Acad. Sci. USA 2017, 114, 2407–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.K. Meralgia paresthetica as a cause of leg discomfort. Can. Med. Assoc. J. 1974, 111, 541–542. [Google Scholar] [PubMed]
- Farber, G.A.; Burks, J.W. Chlorprothixene therapy for herpes zoster neuralgia. South Med. J. 1974, 67, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.W. Chlorprothixene (taractan) in post-herpetic neuralgia and other severe chronic pains. Pain 1978, 5, 367–371. [Google Scholar] [CrossRef]
- Kramer, P.W. The management of postherpetic neuralgia with chlorprothixene. Surg. Neurol. 1981, 15, 102–104. [Google Scholar] [CrossRef]
- Squires, R.F.; Saederup, E. A review of evidence for GABergic predominance/glutamatergic deficit as a common etiological factor in both schizophrenia and affective psychoses: More support for a continuum hypothesis of “functional” psychosis. Neurochem. Res. 1991, 16, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Squires, R.F.; Saederup, E. Clozapine and several other antipsychotic/antidepressant drugs preferentially block the same ‘core’ fraction of GABA(A) receptors. Neurochem. Res. 1998, 23, 1283–1290. [Google Scholar] [CrossRef]
- Roth, B.L.; Craigo, S.C.; Choudhary, M.S.; Uluer, A.; Monsma, F.J., Jr.; Shen, Y.; Meltzer, H.Y.; Sibley, D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994, 268, 1403–1410. [Google Scholar]
- Glusa, E.; Pertz, H.H. Further evidence that 5-HT-induced relaxation of pig pulmonary artery is mediated by endothelial 5-HT(2B) receptors. Br. J. Pharmacol. 2000, 130, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Von Coburg, Y.; Kottke, T.; Weizel, L.; Ligneau, X.; Stark, H. Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett. 2009, 19, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Acs, G.; Palkovits, M.; Blumberg, P.M. Trifluoperazine modulates [3H]resiniferatoxin binding by human and rat vanilloid (capsaicin) receptors and affects 45Ca uptake by adult rat dorsal root ganglion neurones. J. Pharmacol. Exp. Ther. 1995, 274, 1090–1098. [Google Scholar] [PubMed]
- Marchand, F.; Alloui, A.; Chapuy, E.; Hernandez, A.; Pelissier, T.; Ardid, D.; Eschalier, A. The antihyperalgesic effect of venlafaxine in diabetic rats does not involve the opioid system. Neurosci. Lett. 2003, 342, 105–108. [Google Scholar] [CrossRef]
- Aricioğlu, F.; Buldanlioğlu, U.; Salanturoğlu, G.; Ozyalçin, N.S. Evaluation of antinociceptive and anti-inflammatory effects of venlafaxine in the rat. AĞRI 2005, 17, 41–46. [Google Scholar]
- Hajhashemi, V.; Minaiyan, M.; Banafshe, H.R.; Mesdaghinia, A.; Abed, A. The anti-inflammatory effects of venlafaxine in the rat model of carrageenan-induced paw edema. Iran. J. Basic Med. Sci. 2015, 18, 654–658. [Google Scholar] [PubMed]
- Folkesson, A.; Honoré, P.H.; Bjerrum, O.J. Co-administered gabapentin and venlafaxine in nerve injured rats: Effect on mechanical hypersensitivity, motor function and pharmacokinetics. Scand. J. Pain 2010, 1, 91–97. [Google Scholar] [CrossRef]
- Hajhashemi, V.; Banafshe, H.R.; Minaiyan, M.; Mesdaghinia, A.; Abed, A. Antinociceptive effects of venlafaxine in a rat model of peripheral neuropathy: Role of alpha2-adrenergic receptors. Eur. J. Pharmacol. 2014, 738, 230–236. [Google Scholar] [CrossRef]
- Mansouri, M.T.; Naghizadeh, B.; Ghorbanzadeh, B.; Alboghobeish, S.; Amirgholami, N.; Houshmand, G.; Cauli, O. Venlafaxine prevents morphine antinociceptive tolerance: The role of neuroinflammation and the l-arginine-nitric oxide pathway. Exp. Neurol. 2018, 303, 134–141. [Google Scholar] [CrossRef]
- Cegielska-Perun, K.; Tatarkiewicz, J.; Siwek, A.; Dybała, M.; Bujalska-Zadrożny, M. Mechanisms of morphine–venlafaxine interactions in diabetic neuropathic pain model. Pharmacol. Rep. 2015, 67, 90–96. [Google Scholar] [CrossRef]
- Rowbotham, M.C.; Goli, V.; Kunz, N.R.; Lei, D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: A double-blind, placebo-controlled study. Pain 2004, 110, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.Y.; Li, Q.F.; Song, D.P.; An, Z.M.; Liu, Y.P.; Ran, X.W.; Wu, R.H.; Tian, H.M. Effect of venlafaxine and carbamazepine for painful peripheral diabetic neuropathy: A randomized, double-blind and double-dummy, controlled multi-center trial. Chin. J. Evid. Based Med. 2006, 6, 321–327. [Google Scholar]
- Piletz, J.E.; Halaris, A.; Iqbal, O.; Hoppensteadt, D.; Fareed, J.; Zhu, H.; Sinacore, J.; Devane, C.L. Pro-inflammatory biomakers in depression: Treatment with venlafaxine. World J. Biol. Psychiatry 2009, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, J.H.; Min, B.-H.; Lee, J.H.; Son, H.J.; Kim, J.J.; Rhee, P.-L. Efficacy of Venlafaxine for Symptomatic Relief in Young Adult Patients with Functional Chest Pain: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. Am. J. Gastroenterol. 2010, 105, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Razazian, N.; Baziyar, M.; Moradian, N.; Afshari, D.; Bostani, A.; Mahmoodi, M. Evaluation of the efficacy and safety of pregabalin, venlafaxine, and carbamazepine in patients with painful diabetic peripheral neuropathy. A randomized, double-blind trial. Neurosciences (Riyadh) 2014, 19, 192–198. [Google Scholar] [PubMed]
- Gallagher, H.C.; Gallagher, R.M.; Butler, M.; Buggy, D.J.; Henman, M.C. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst. Rev. 2015, 2015, CD011091. [Google Scholar] [CrossRef] [PubMed]
- Farshchian, N.; Alavi, A.; Heydarheydari, S.; Moradian, N. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother. Pharmacol. 2018, 82, 787–793. [Google Scholar] [CrossRef]
- Raabe, R.; Gentile, L. Antidepressant interactions with the NMDA NR1-1b subunit. J. Biophys. 2008, 2008, 474205. [Google Scholar] [CrossRef]
- Yilmaz, N.; Demirdas, A.; Yilmaz, M.; Sutcu, R.; Kirbas, A.; Cure, M.C.; Eren, I. Effects of venlafaxine and escitalopram treatments on NMDA receptors in the rat depression model. J. Membr. Biol. 2011, 242, 145–151. [Google Scholar] [CrossRef]
- Tamási, V.; Petschner, P.; Adori, C.; Kirilly, E.; Ando, R.D.; Tothfalusi, L.; Juhasz, G.; Bagdy, G. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also Glutamatergic- and insulin-mediated neuronal processes. PLoS ONE 2014, 9, e113662. [Google Scholar] [CrossRef]
- Khalifa, M.; Daleau, P.; Turgeon, A.J. Mechanism of sodium channel block by venlafaxine in guinea pig ventricular myocytes. J. Pharmacol. Exp. Ther. 1999, 291, 280–284. [Google Scholar] [PubMed]
- Schreiber, S.; Backer, M.M.; Pick, C.G. The antinociceptive effect of venlafaxine in mice is mediated through opioid and adrenergic mechanisms. Neurosci. Lett. 1999, 273, 85–88. [Google Scholar] [CrossRef]
- Sikka, P.; Kaushik, S.; Kumar, G.; Kapoor, S.; Bindra, V.K.; Saxena, K.K. Study of antinociceptive activity of SSRI (fluoxetine and escitalopram) and atypical antidepressants (venlafaxine and mirtazepine) and their interaction with morphine and naloxone in mice. J. Pharm. Bioallied. Sci. 2011, 3, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Dreshfield-Ahmad, L.J.; Threlkeld, P.G.; Shaw, J.L.; Thompson, L.; Nelson, D.L.; Hemrick-Luecke, S.K.; Wong, D.T. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology 2001, 25, 871–880. [Google Scholar] [CrossRef]
- Miskovic, M. Comparison of tolerance of venlafaxine, paroxetine and amitriptyline in depression therapy. Med. Arch. 2015, 69, 107–109. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, C.; Mao, Y. Determination of venlafaxine and its active metabolite O-desmethylvenlafaxine in human plasma by HPLC fluorescence. Gen. Psychiatr. 2018, 31, e000010. [Google Scholar] [CrossRef]
- Miyano, K.; Minami, K.; Yokoyama, T.; Ohbuchi, K.; Yamaguchi, T.; Murakami, S.; Shiraishi, S.; Yamamoto, M.; Matoba, M.; Uezono, Y. Tramadol and its metabolite m1 selectively suppress transient receptor potential ankyrin 1 activity, but not transient receptor potential vanilloid 1 activity. Anesth. Analg. 2015, 120, 790–798. [Google Scholar] [CrossRef]
- Plummer, N.W.; Meisler, M.H. Evolution and diversity of mammalian sodium channel genes. Genomics 1999, 57, 323–331. [Google Scholar] [CrossRef]
- Brouwer, B.A.; Merkies, I.S.; Gerrits, M.M.; Waxman, S.G.; Hoeijmakers, J.G.; Faber, C.G. Painful neuropathies: The emerging role of sodium channelopathies. J. Peripher. Nerv. Syst. 2014, 19, 53–65. [Google Scholar] [CrossRef]
- Lauria, G.; Ziegler, D.; Malik, R.; Merkies, I.S.; Waxman, S.G.; Faber, C.G.; PROPANE Study Group. The role of sodium channels in painful diabetic and idiopathic neuropathy. Curr. Diab. Rep. 2014, 14, 538. [Google Scholar] [CrossRef]
- Rasakham, K.; Liu-Chen, L.Y. Sex differences in kappa opioid pharmacology. Life Sci. 2011, 88, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Chartoff, E.H.; Mavrikaki, M. Sex Differences in Kappa Opioid Receptor Function and Their Potential Impact on Addiction. Front. Neurosci. 2015, 9, 466. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.W.; Ratnaseelan, A.M.; Veenema, A.H. Robust age, but limited sex, differences in mu-opioid receptors in the rat brain: Relevance for reward and drug-seeking behaviors in juveniles. Brain Struct. Funct. 2018, 223, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Dance, A. Why the sexes don’t feel pain the same way. Nature 2019, 567, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.; Norton, J.; Goodwillie, A.; Pfaff, D.W. Sex differences in mouse behavior following pyrilamine treatment: Role of histamine 1 receptors in arousal. Pharmacol. Biochem. Behav. 2004, 79, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Granados-Soto, V.; Alonso-López, R.; Asomoza-Espinosa, R.; Rufino, M.O.; Gomes-Lopes, L.D.; Ferreira, S.H. Participation of COX, IL-1 beta and TNF alpha in formalin-induced inflammatory pain. Proc. West Pharmacol. Soc. 2001, 44, 15–17. [Google Scholar] [PubMed]
- Chichorro, J.G.; Lorenzetti, B.B.; Zampronio, A.R. Involvement of bradykinin, cytokines, sympathetic amines and prostaglandins in formalin-induced orofacial nociception in rats. Br. J. Pharmacol. 2004, 141, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. The role of anti-inflammatory treatment in psychiatric disorders. Psychiatr. Danub. 2013, 25, 292–298. [Google Scholar]
- Bai, Y.M.; Chiou, W.F.; Su, T.P.; Li, C.T.; Chen, M.H. Pro-inflammatory cytokine associated with somatic and pain symptoms in depression. J. Affect. Disord. 2014, 155, 28–34. [Google Scholar] [CrossRef]
- Post, R.M.; Uhde, T.W.; Roy-Byrne, P.P.; Joffe, R.T. Antidepressant effects of carbamazepine. Am. J. Psychiatry 1986, 143, 29–34. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Analgesic Effect: Animal Models | Analgesic Effect: Clinical Trial | Antipruritic Effect: Animal Models | Antipruritic Effect: Clinical Trial | Dosage in Clinical Trials, Literature Data (Our Study) | Main Side Effects Led to Withdrawals and Refusal of Treatment |
|---|---|---|---|---|---|---|
| Tianeptine | formalin test; mechanical allodynia; fibromyalgia | chronic pain in patients with chronic renal failure (CRF) | ND | neuropathic pruritus in male patients with CRF | 20–40 mg/day (37.5 mg/day) | ND * |
| Citalopram | hot plate test; the second phase of the formalin test; thermal hypersensitivity in the chronic constriction injury (CCI) model | psychosomatic pain; diabetic neuropathy (mild effect); migraine (presumably); neuropathic pain in patients with CRF (mild effect) | ND | itching in psoriasis (mild effect); neuropathic pruritus in patients with CRF (mild effect) | 20–40 mg/day (20 mg/day) | nausea, vomiting, epileptic seizures, tachycardia, dizziness, gastric upset, diarrhea |
| Mianserin | hot plate test (enhancement of metamizol); tail flick test (enhancement of metamizole and indometacin); diabetic neuropathy | neuropathic pain in patients with CRF; tension headache; chronic pain associated with “true” depression | potentiation of hindlimb scratching | neuropathic pruritus in male patients with CRF | 30–90 mg/day (30 mg/day) | lack of appetite, dryness of mouth, thirst, drowsiness, dullness |
| Carbamazepine | hot plate test; inflammatory pain; trigeminal neuralgia; chemotherapy-induced painful neuropathy | diabetic neuropathy; trigeminal neuralgia | ND | itching in sclerosis; brachioodal itch; trigeminal trophic syndrome | 200–800 mg/day (300 mg/day) | giddiness, rash, sleeplessness |
| Trazodone | hot plate test; thermal hyperalgesia; formalin induced pain | diabetic neuropathy; adjuvant therapy in fibromyalgia | ND | diffuse pruritus | 50–300 mg/day (50–100 mg/day) | dizziness, headache, tachycardia, stomachache |
| Chlorprothixene | anesthesia of experimental animals | postherpetic neuralgia; adjuvant therapy in meralgia paraesthetica | ND | diffuse pruritus | 50–400 mg/day (15 mg/day) | psychical abnormalities, drowsiness, vertigo |
| Venlafaxine | hyperalgesia in the CCI model; carrageenan-induced pain and inflammation; mechanical hypersensitivity induced by SNI; neuroinflammation and oxidative stress | diabetic neuropathy; chemotherapy-induced peripheral neuropathy; visceral hyperalgesia; inflammation | ND | ND | 37.5–225 mg/day | nausea, somnolence, headache, insomnia, sexual dysfunction, dizziness |
| Drug | NMDA (inhib.) | AMPA (inhib.) | Sodium (inhib.) | Calcium (inhib.) | GABA(A) (activ.) | Opioid (activ.) | Cannabinoid (modul.) | 5HT7 (activ.) | NCX (inhib.) | Histamine (inhib.) | TRP (inhib.) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tianeptine | − | − | ND | − | − | + | ND | + | ND | − | ND |
| Citalopram | − | − | weak | + | − | − | + | − | ND | + | ND |
| Mianserin | − | − | + | there are + and − | − | + | ND | − | ND | + | ND |
| Carbamazepine | − | − | + | there are + and − | + | ND | possible | ND | ND | ND | − |
| Trazodone | − | ND | there are + and − | + | + | + | ND | + | ND | + | ND |
| Chlorprothixene | + | ND | + | ND | − | ND | ND | − | ND | + | possible |
| Venlafaxine | − | ND | + | ND | ND | + | ND | − | ND | − | possible |
| Pain (Dartmouth Pain Questionnaire) | ||||
| Drug | Men | Women | ||
| Before Treatment | After Treatment | Before Treatment | After Treatment | |
| tianeptine | 0.44 ± 0.21 | 0.21 ± 0.16 | 0.91 ± 0.16 | 0.45 ± 0.21 |
| citalopram | 0.78 ± 0.25 | 0.64 ± 0.18 | 0.85 ± 0.16 | 0.66 ± 0.12 |
| mianserin | 0.81 ± 0.23 | 0.27 ± 0.06 | 0.93 ± 0.18 | 0.71 ± 0.16 |
| comparison group | 0.13 ± 0.07 | 0.13 ± 0.1 | 0.23 ± 0.12 | 0.22 ± 0.11 |
| Itch, Scoring Atopic Dermatitis index scale (SCORAD) | ||||
| Drug | Men | Women | ||
| Before Treatment | After Treatment | Before Treatment | After Treatment | |
| tianeptine | 17.4 ± 8.1 | 10.8 ± 5.6 | 15.5 ± 1.3 | 12.5 ± 1.7 |
| citalopram | 16.45 ± 6.7 | 14.45 ± 6.0 | 13.75 ± 1.8 | 10.75 ± 1.3 |
| mianserin | 18 ± 7.5 | 4.75 ± 1.8 | 8.41 ± 2.1 | 6.97 ± 1.3 |
| comparison group | 9.16 ± 3.44 | 9.42 ± 3.6 | 7.64 ± 1.8 | 7.96 ± 1.6 |
| Patients | Pain | Itch | Depression | Anxiety |
|---|---|---|---|---|
| men | 76.1% | 84.0% | 20.9% | 31.1% |
| women | 63.1% | 44.0% | 18.9% | 30.0% |
| Drugs | Receptors | Type of Analgesic/Antipruritic Effect (Animal Models or/and Clinical Studies) |
|---|---|---|
| Trazodone chlorprothixene carbamazepine mianserin | voltage-dependent sodium channels in the peripheral nervous system | diabetic neuropathy, postherpetic neuralgia |
| citalopram mianserin trazodone venlafaxine | cannabinoid or opioid receptors | supraspinal analgesia |
| mianserin | κ-opioid receptors | sex-dependent analgesia |
| tianeptine | μ-opioid receptors | sex-independent analgesia |
| tianeptine citalopram trazodone venlafaxine carbamazepine | no similarities in pharmacological profiles found | inflammatory pain/antidepressants (or has antidepressant action) |
| chlorprothixene | histamine receptors + possible TRP channel * | sex-independent antipruritic action |
| mianserin carbamazepine tianeptine | no similarities in pharmacological profiles found | sex-dependent antipruritic action |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belinskaia, D.A.; Belinskaia, M.A.; Barygin, O.I.; Vanchakova, N.P.; Shestakova, N.N. Psychotropic Drugs for the Management of Chronic Pain and Itch. Pharmaceuticals 2019, 12, 99. https://doi.org/10.3390/ph12020099
Belinskaia DA, Belinskaia MA, Barygin OI, Vanchakova NP, Shestakova NN. Psychotropic Drugs for the Management of Chronic Pain and Itch. Pharmaceuticals. 2019; 12(2):99. https://doi.org/10.3390/ph12020099
Chicago/Turabian StyleBelinskaia, Daria A., Mariia A. Belinskaia, Oleg I. Barygin, Nina P. Vanchakova, and Natalia N. Shestakova. 2019. "Psychotropic Drugs for the Management of Chronic Pain and Itch" Pharmaceuticals 12, no. 2: 99. https://doi.org/10.3390/ph12020099