Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis


{kind=link}
Abstract
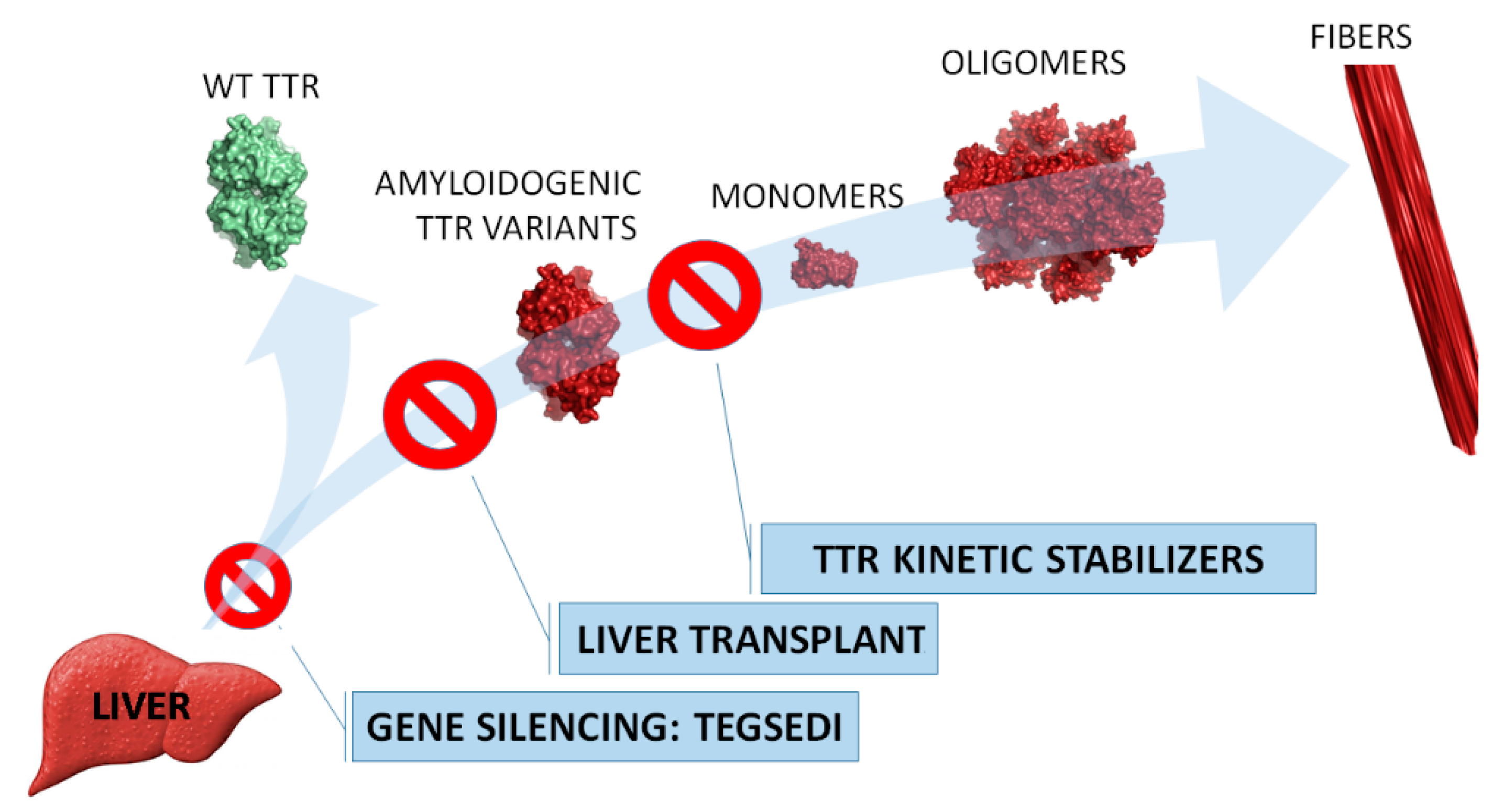
:1. Introduction
2. Tegsedi (Inotersen)
2.1. Name
2.2. Uses
2.3. Mechanism of Action
2.4. Clinical Studies
3. Perspectives
Funding
Conflicts of Interest
References
- Blake, C.C.F.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Mascarenhas Saraiva, M.J.; Birken, S.; Costa, P.P.; Goodman, D.S. Amyloid fibril protein in familial amyloidotic polyneuropathy, Portuguese type. Definition of molecular abnormality in transthyretin (prealbumin). J. Clin. Invest. 1984, 74, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Suhr, O.B.; Hund, E.; Obici, L.; Tournev, I.; Campistol, J.M.; Slama, M.S.; Hazenberg, B.P.; Coelho, T.; European Network for, T.-F. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr. Opin. Neurol. 2016, 29, S14–S26. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with Hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018, 14, 570. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Ann. Rev. Med. 2019, 70, 307–321. [Google Scholar]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, E.J.; Guo, S.; Benson, M.D.; Booten, S.; Freier, S.; Hughes, S.G.; Kim, T.W.; Jesse Kwoh, T.; Matson, J.; Norris, D.; et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016, 23, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.R.; Gales, L.; Damas, A.M.; Cardoso, I.; Saraiva, M.J. Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 587–596. [Google Scholar] [CrossRef]
- Gales, L.; Aronson, J.K. Thyroid hormones, iodine and iodides, and antithyroid drugs. In Side Effects of Drugs Annual; Elsevier: Amsterdam, Netherlands, 2014; Volume 35, pp. 747–761. [Google Scholar]
- Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. PNAS 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Gonçalves, N.P.; Saraiva, M.J.; Almeida, M.R. Curcumin: A multi-Target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 2016, 6, 26623. [Google Scholar] [CrossRef] [PubMed]
- Maia, F.; Almeida, M.D.R.; Gales, L.; Kijjoa, A.; Pinto, M.M.M.; Saraiva, M.J.; Damas, A.M. The binding of xanthone derivatives to transthyretin. Biochem. Pharmacol. 2005, 70, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA—J. Am. Med. Assoc. 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.R.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef]
- Gales, L.; Almeida, M.R.; Arsequell, G.; Valencia, G.; Saraiva, M.J.; Damas, A.M. Iodination of salicylic acid improves its binding to transthyretin. BBA-Proteins Proteom. 2008, 1784, 512–517. [Google Scholar] [CrossRef]
- Gales, L.; Macedo-Ribeiro, S.; Arsequell, G.; Valencia, G.; Saraiva, M.J.; Damas, A.M. Human transthyretin in complex with iododiflunisal: Structural features associated with a potent amyloid inhibitor. Biochem. J. 2005, 388, 615–621. [Google Scholar] [CrossRef]
- Mairal, T.; Nieto, J.; Pinto, M.; Almeida, M.R.; Gales, L.; Ballesteros, A.; Barluenga, J.; Pérez, J.J.; Vázquez, J.T.; Centeno, N.B.; et al. Iodine atoms: A new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS ONE 2009, 4, e4124. [Google Scholar] [CrossRef]
- Ferreira, N.; Pereira-Henriques, A.; Attar, A.; Klärner, F.G.; Schrader, T.; Bitan, G.; Gales, L.; Saraiva, M.J.; Almeida, M.R. Molecular Tweezers Targeting Transthyretin Amyloidosis. Neurotherapeutics 2014, 11, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Gales, L.; Saraiva, M.J.; Damas, A.M. Structural basis for the protective role of sulfite against transthyretin amyloid formation. BBA-Proteins Proteom. 2007, 1774, 59–64. [Google Scholar] [CrossRef]
- Yokoyama, T.; Mizuguchi, M. Crown Ethers as Transthyretin Amyloidogenesis Inhibitors. J. Med. Chem. 2019, 62, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Baker, B.F.; Witztum, J.L.; Kwoh, T.J.; Pham, N.C.; Salgado, N.; McEvoy, B.W.; Cheng, W.; Hughes, S.G.; Bhanot, S.; et al. The Effects of 2′-O-Methoxyethyl Containing Antisense Oligonucleotides on Platelets in Human Clinical Trials. Nucleic Acid Ther. 2017, 27, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Kincaid, J.C.; Dyck, P.J.B.; Chaudhry, V.; Goyal, N.A.; Alves, C.; Salhi, H.; Wiesman, J.F.; Labeyrie, C.; Robinson-Papp, J.; et al. Assessing mNIS+7 Ionis and international neurologists’ proficiency in a familial amyloidotic polyneuropathy trial. Muscle Nerve 2017, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Peripheral Nerve, S. Diabetic polyneuropathy in controlled clinical trials: Consensus report of the peripheral nerve society. Ann. Neurol. 1995, 38, 478–482. [Google Scholar] [CrossRef]
- Vinik, E.J.; Vinik, A.I.; Paulson, J.F.; Merkies, I.S.J.; Packman, J.; Grogan, D.R.; Coelho, T. Norfolk QOL-DN: Validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J. Peripher. Nerv. Syst. 2014, 19, 104–114. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gales, L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals 2019, 12, 78. https://doi.org/10.3390/ph12020078
Gales L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals. 2019; 12(2):78. https://doi.org/10.3390/ph12020078
Chicago/Turabian StyleGales, Luís. 2019. "Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis" Pharmaceuticals 12, no. 2: 78. https://doi.org/10.3390/ph12020078