Iron Supplementation Therapy, A Friend and Foe of Mycobacterial Infections?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. General Context
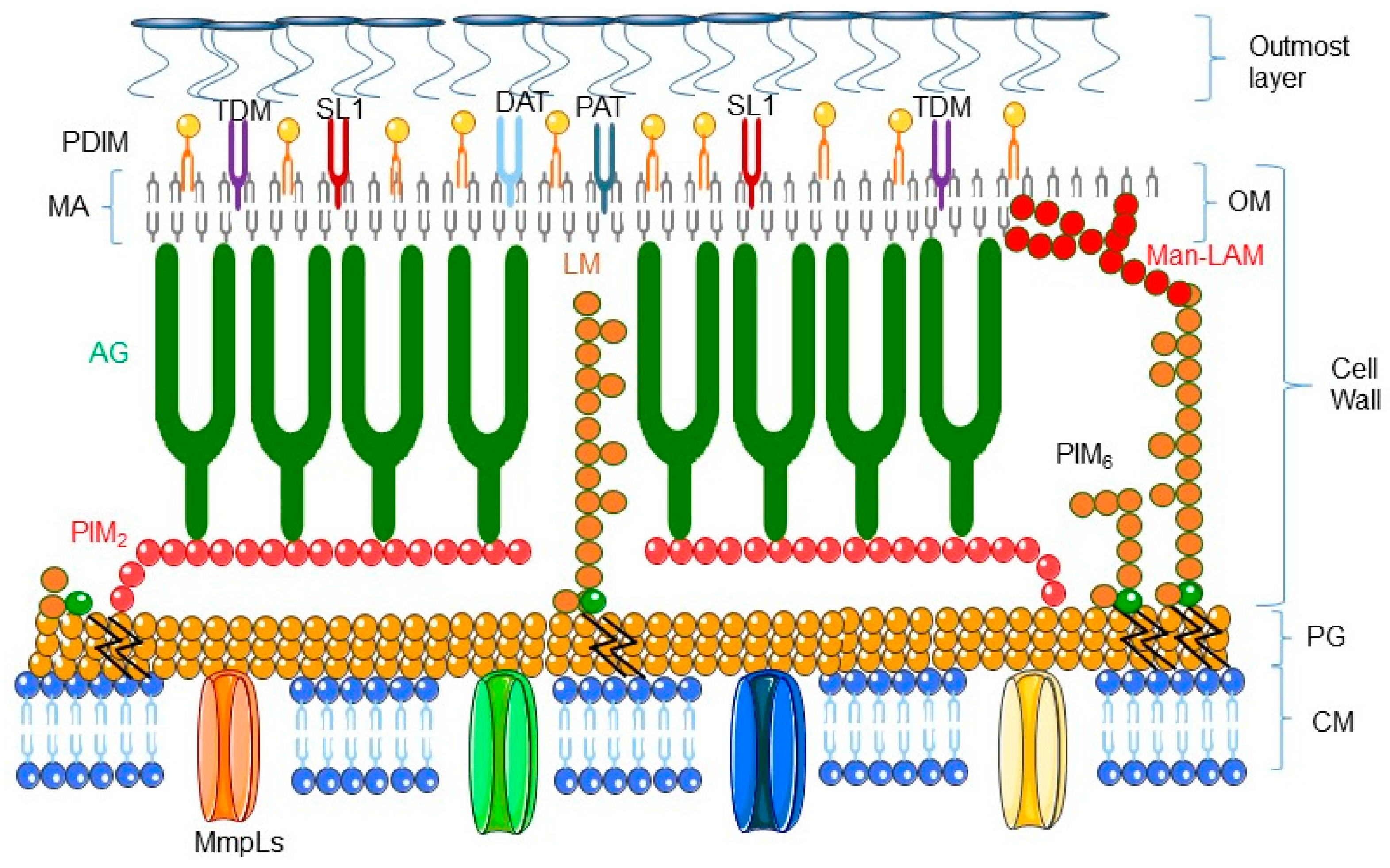
2. Mycobacteria: The Smart Pathogens
2.1. Host Response to Mycobacteria Through Immune Cell Activation
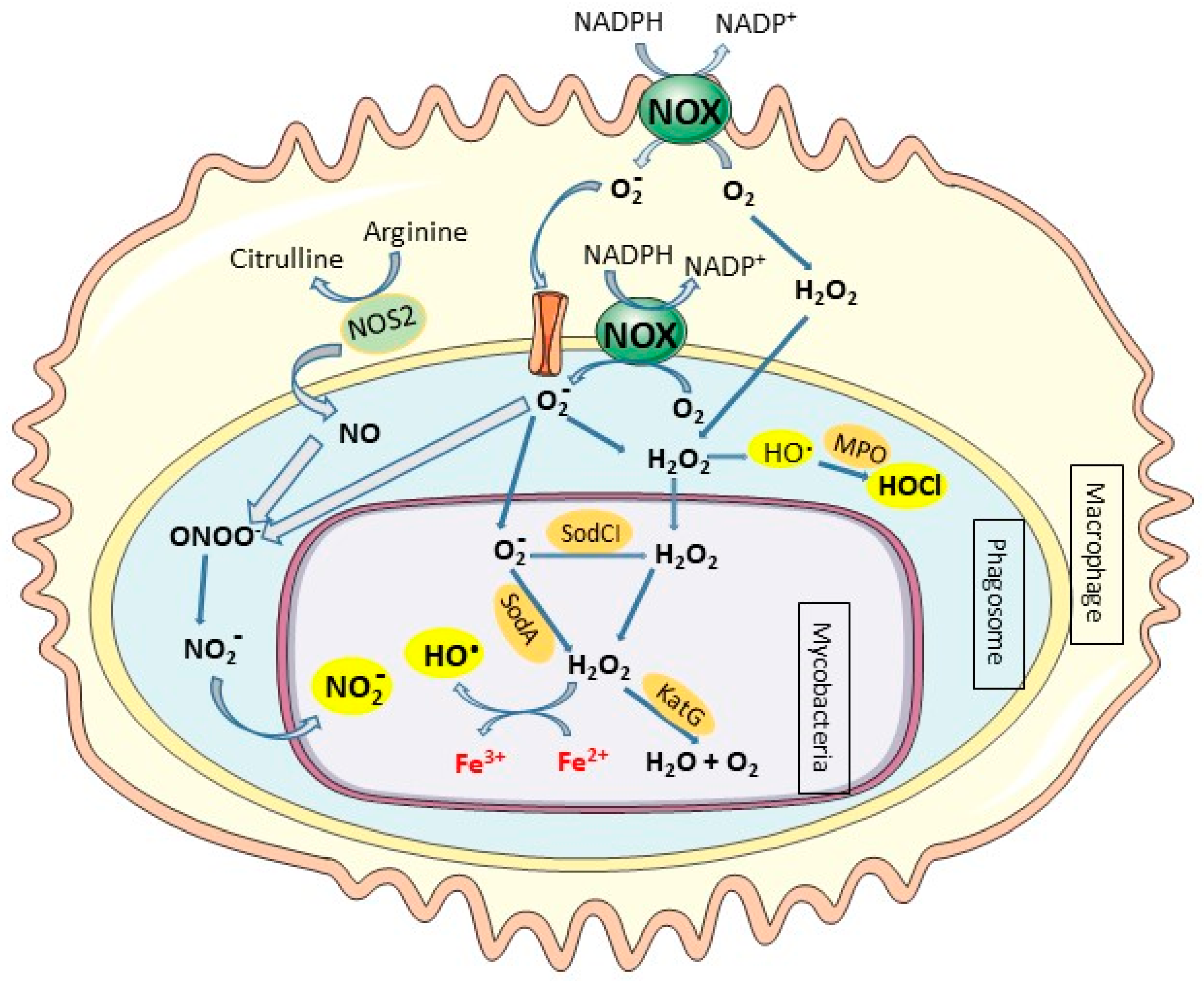
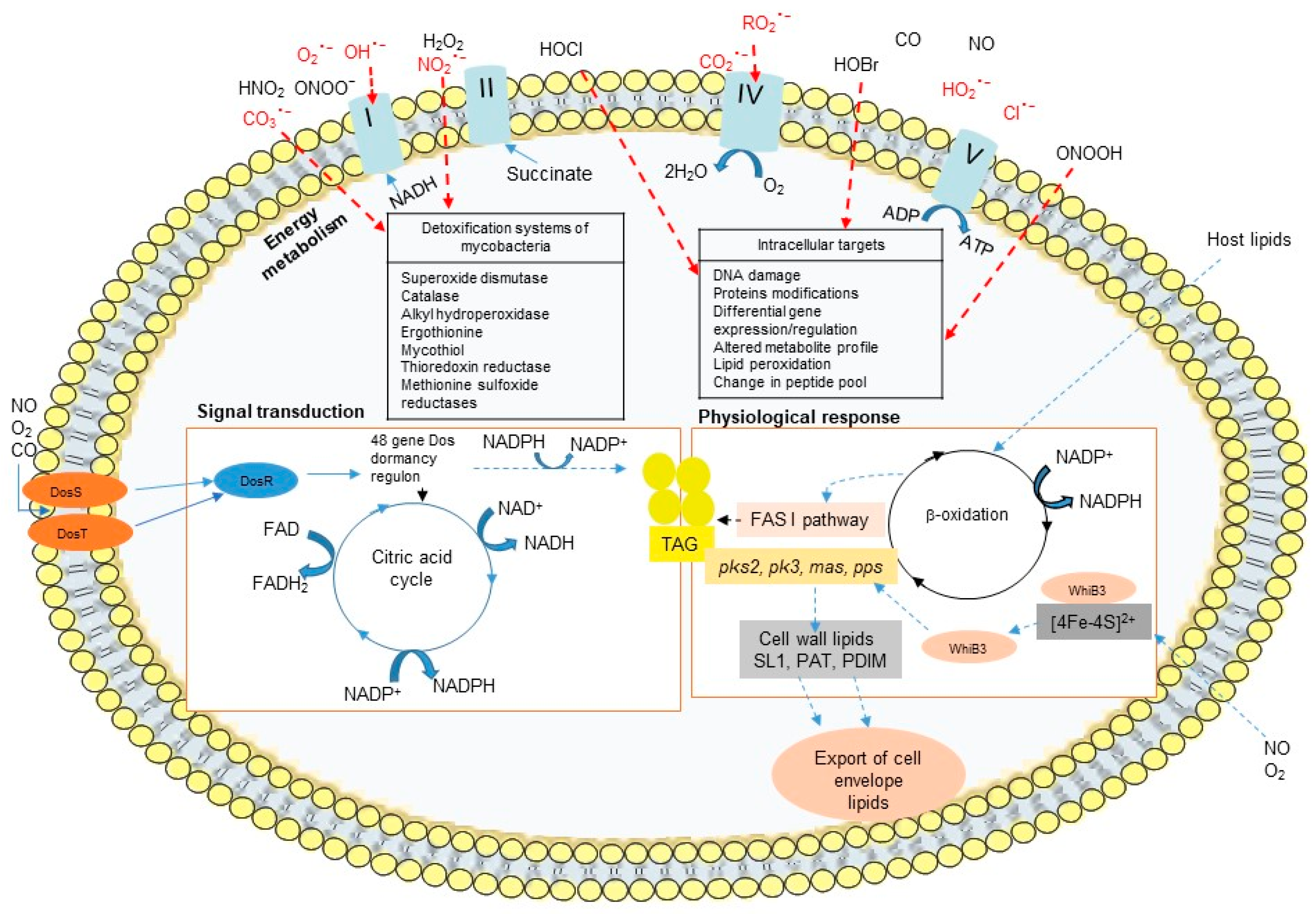
2.2. Host Response to Mycobacteria Through Oxidative Stress Induction: A Critical Role of Iron
3. The Iron Tug-of-War Between Host and Mycobacteria
3.1. The Importance of Iron for Mycobacteria
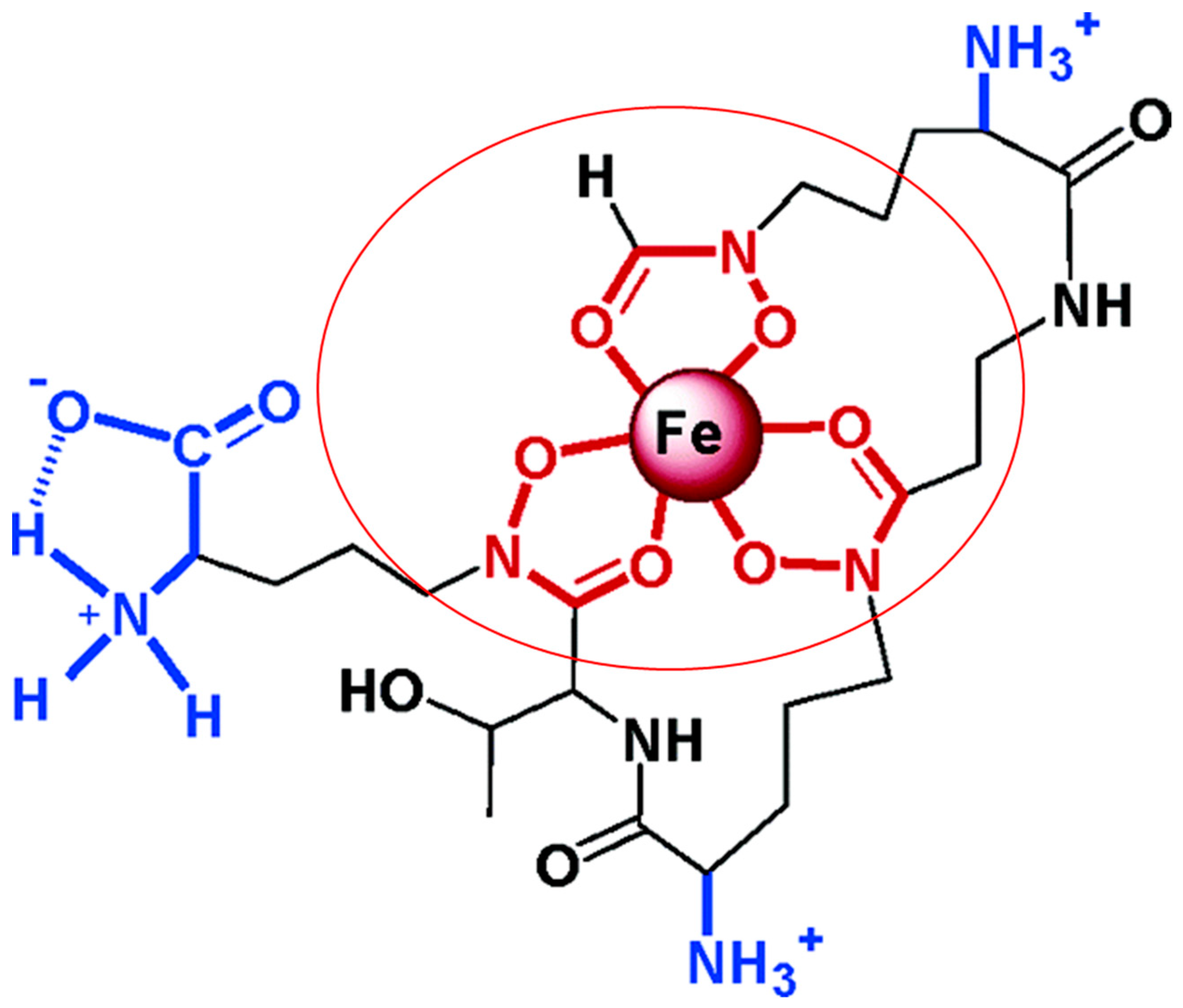
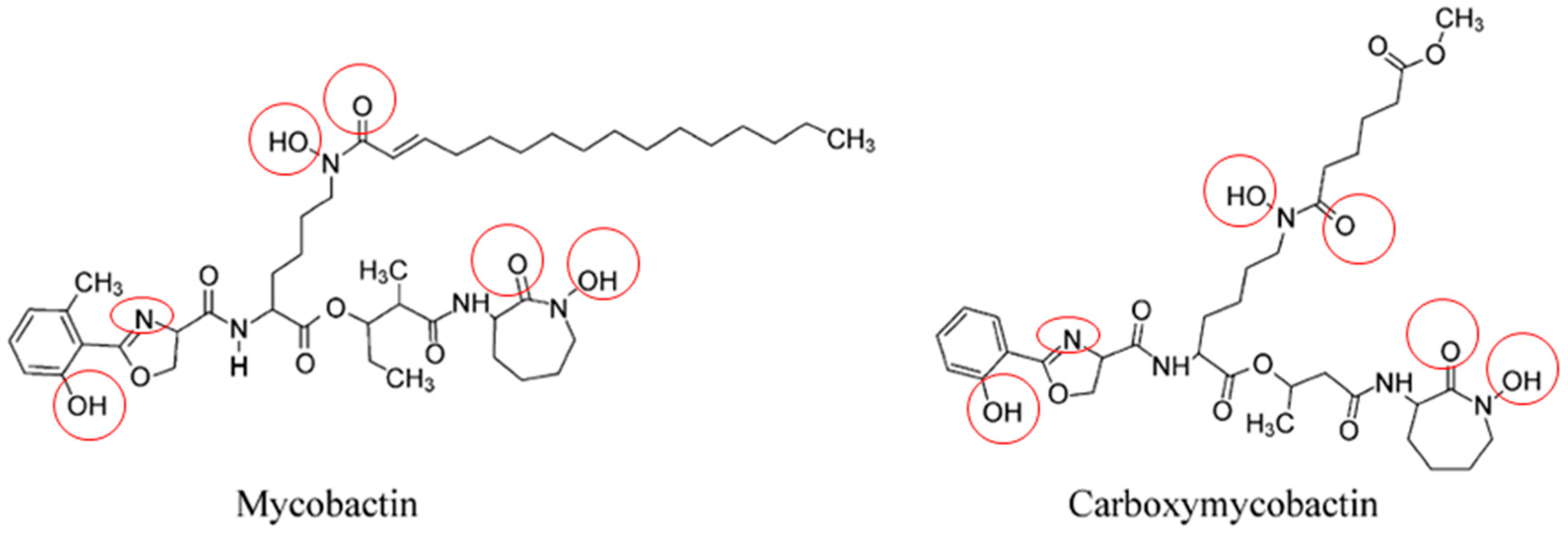
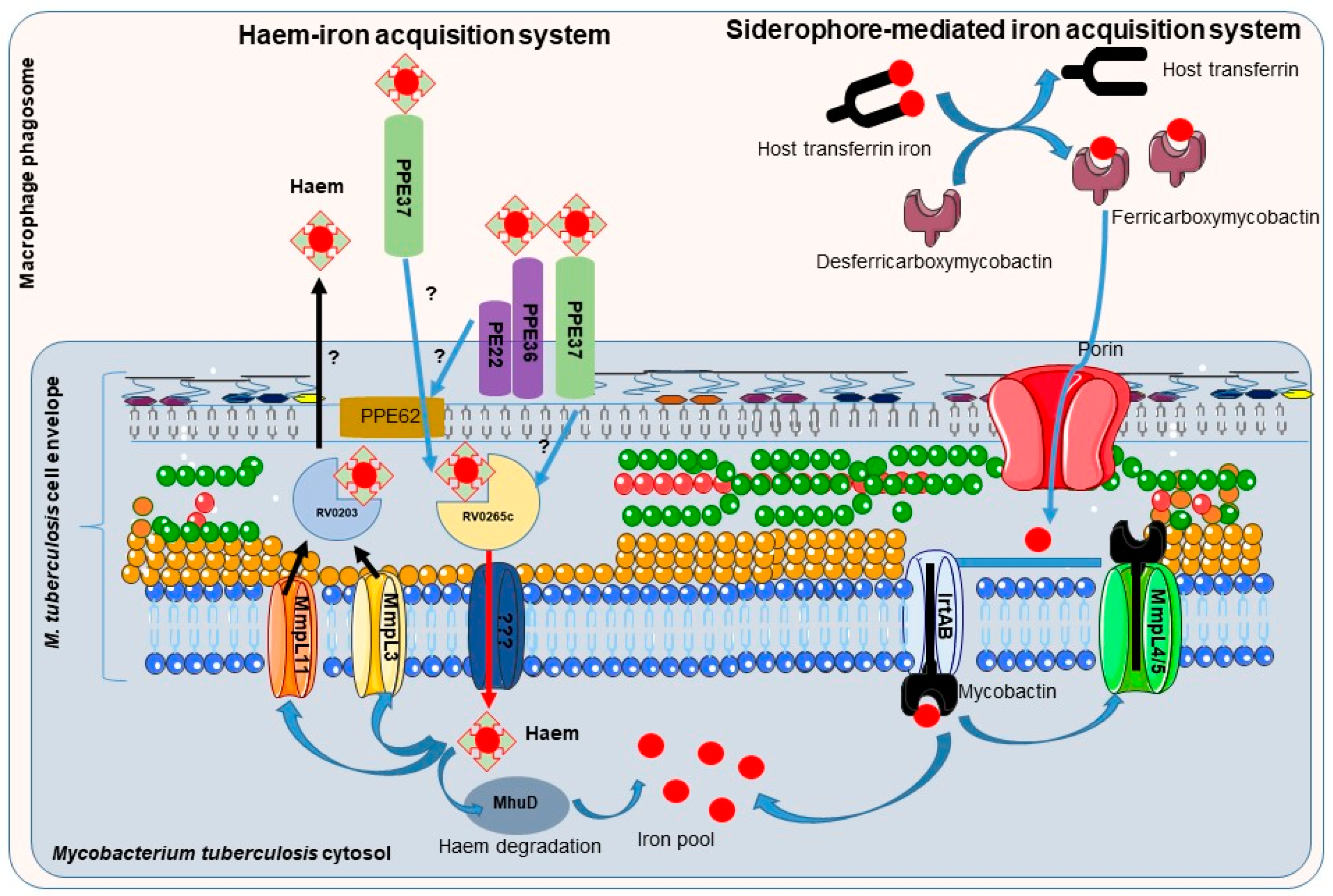
3.1.1. Siderophores: The Mycobacteria Iron Scavengers
3.1.2. Mycobacteria Heme-Iron Acquisition Systems
3.2. Mycobacteria Protection against the Harmful Effects of Iron, ROS and RNS
3.2.1. Protection against Excess of Iron
3.2.2. Antioxidant Systems in Mycobacteria
4. Implication of Iron in the Host Arsenal Defense against Mycobacterial Infection
4.1. Host Cellular Iron Metabolism during Mycobacteria Infection
4.2. Systemic Iron Homeostasis during Mycobacterial Infections
4.3. Iron and Antimicrobial Peptides
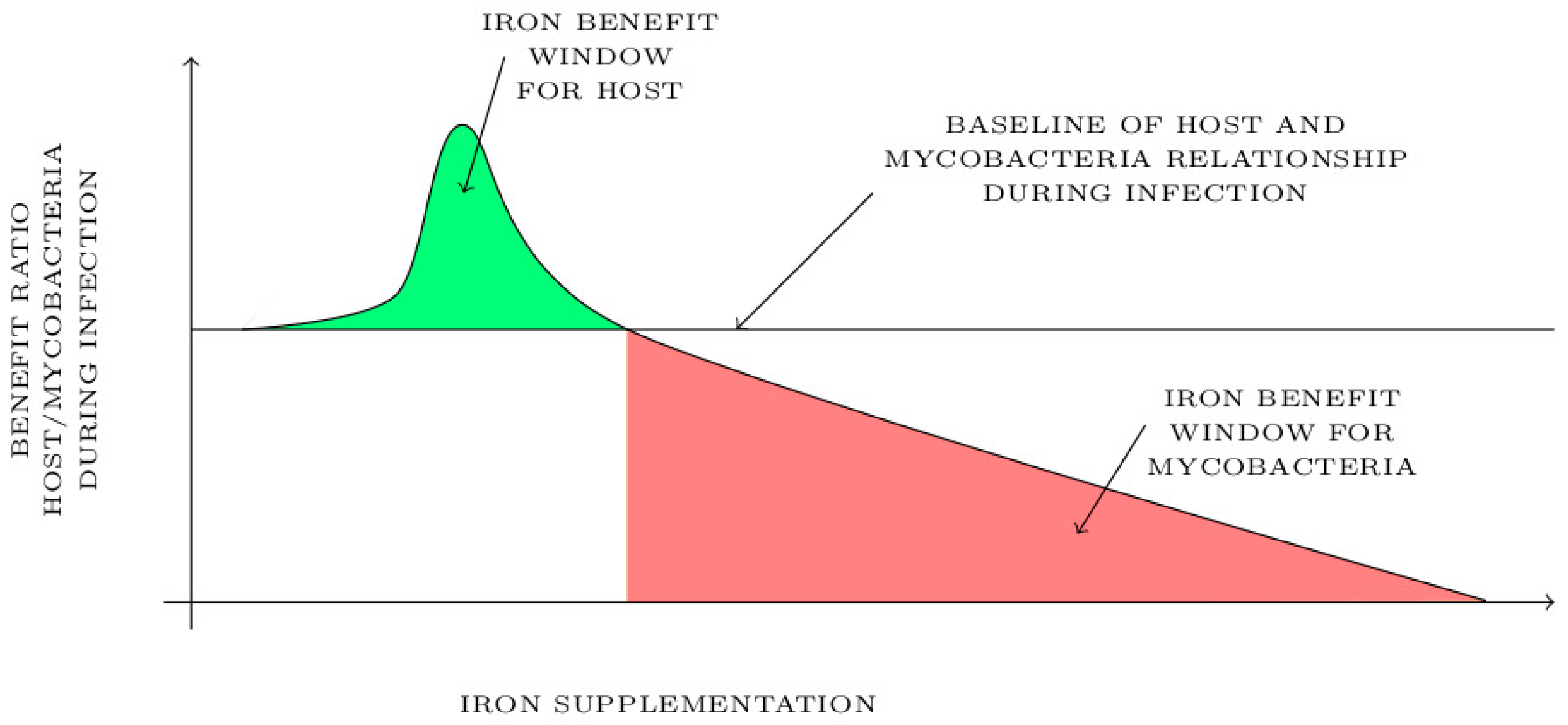
5. Future Therapeutic Use of Iron in Mycobacterial Infections: Iron, a Friend or Foe
5.1. Investigation of the Effect of Iron on Mycobacterial Growth in Host Cell-Free Culture
5.2. Investigation of the Effect of Iron on Mycobacterial Growth in Macrophages
5.3. Investigation of the Effect of Iron on Mycobacterial Growth In Vivo
5.3.1. Investigation of the Effect of Iron on Mycobacterial Growth in Mice
5.3.2. Investigation of the Effect of Iron on Mycobacterial Growth in Human
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Behr, M.A. Building a better bacillus: The emergence of Mycobacterium tuberculosis. Front. Microbiol. 2014, 5, 139. [Google Scholar] [CrossRef]
- Queiroz, A.; Riley, L.W. Bacterial immunostat: Mycobacterium tuberculosis lipids and their role in the host immune response. Rev. Soc. Bras. Med. Trop. 2017, 50, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosma, C.L.; Sherman, D.R.; Ramakrishnan, L. The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 2003, 57, 641–676. [Google Scholar] [CrossRef]
- Larsson, L.O.; Polverino, E.; Hoefsloot, W.; Codecasa, L.R.; Diel, R.; Jenkins, S.G.; Loebinger, M.R. Pulmonary disease by non-tuberculous mycobacteria—Clinical management, unmet needs and future perspectives. Expert Rev. Respir. Med. 2017, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Ramakrishnan, L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009, 136, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Ramakrishnan, L. “The very pulse of the machine”: The tuberculous granuloma in motion. Immunity 2008, 28, 146–148. [Google Scholar] [CrossRef]
- Korbel, D.S.; Schneider, B.E.; Schaible, U.E. Innate immunity in tuberculosis: Myths and truth. Microbes Infect. 2008, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.F.; Hibbs, J.B., Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991, 3, 65–70. [Google Scholar] [CrossRef]
- MacMicking, J.D.; North, R.J.; LaCourse, R.; Mudgett, J.S.; Shah, S.K.; Nathan, C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Scanga, C.A.; Mohan, V.P.; Tanaka, K.; Alland, D.; Flynn, J.L.; Chan, J. The inducible nitric oxide synthase locus confers protection against aerogenic challenge of both clinical and laboratory strains of Mycobacterium tuberculosis in mice. Infect. Immun. 2001, 69, 7711–7717. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Allen, L.A.; Apicella, M.; Nauseef, W.M. NADPH oxidase activation and assembly during phagocytosis. J. Immunol. 1999, 163, 6732–6740. [Google Scholar] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, J.; Saunders, B.M.; Appelberg, R.; Orme, I.M.; Silva, M.T.; Cooper, A.M. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect. Immun. 2000, 68, 577–583. [Google Scholar] [CrossRef]
- Martineau, A.R.; Newton, S.M.; Wilkinson, K.A.; Kampmann, B.; Hall, B.M.; Nawroly, N.; Packe, G.E.; Davidson, R.N.; Griffiths, C.J.; Wilkinson, R.J. Neutrophil-mediated innate immune resistance to mycobacteria. J. Clin. Investig. 2007, 117, 1988–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Aguilar, M.D.; Arce-Paredes, P.; Aquino-Vega, M.; Rodriguez-Martinez, S.; Rojas-Espinosa, O. Fate of Mycobacterium tuberculosis in peroxidase-loaded resting murine macrophages. Int. J. Mycobacteriol. 2013, 2, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef] [Green Version]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency: Prevalence and clinical significance. Ann. Intern. Med. 1981, 95, 293–301. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.; Rogers, H.J.; Bullen, J.J. Iron, plasmids and infection. Nature 1980, 284, 508–509. [Google Scholar] [CrossRef]
- Hamilton, T.A.; Gray, P.W.; Adams, D.O. Expression of the transferrin receptor on murine peritoneal macrophages is modulated by in vitro treatment with interferon gamma. Cell. Immunol. 1984, 89, 478–488. [Google Scholar] [CrossRef]
- Marakalala, M.J.; Raju, R.M.; Sharma, K.; Zhang, Y.J.; Eugenin, E.A.; Prideaux, B.; Daudelin, I.B.; Chen, P.Y.; Booty, M.G.; Kim, J.H.; et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat. Med. 2016, 22, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurthkoti, K.; Amin, H.; Marakalala, M.J.; Ghanny, S.; Subbian, S.; Sakatos, A.; Livny, J.; Fortune, S.M.; Berney, M.; Rodriguez, G.M. The Capacity of Mycobacterium tuberculosis To Survive Iron Starvation Might Enable It To Persist in Iron-Deprived Microenvironments of Human Granulomas. mBio 2017, 8, e01092-17. [Google Scholar] [CrossRef] [PubMed]
- Olakanmi, O.; Schlesinger, L.S.; Ahmed, A.; Britigan, B.E. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-gamma and hemochromatosis. J. Biol. Chem. 2002, 277, 49727–49734. [Google Scholar] [CrossRef]
- Sharman, G.J.; Williams, D.H.; Ewing, D.F.; Ratledge, C. Isolation, purification and structure of exochelin MS, the extracellular siderophore from Mycobacterium smegmatis. Biochem. J. 1995, 305 Pt 1, 187–196. [Google Scholar] [CrossRef]
- Sharman, G.J.; Williams, D.H.; Ewing, D.F.; Ratledge, C. Determination of the structure of exochelin MN, the extracellular siderophore from Mycobacterium neoaurum. Chem. Biol. 1995, 2, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Dhungana, S.; Ratledge, C.; Crumbliss, A.L. Iron chelation properties of an extracellular siderophore exochelin MS. Inorg. Chem. 2004, 43, 6274–6283. [Google Scholar] [CrossRef] [PubMed]
- Barclay, R.; Ratledge, C. Mycobactins and exochelins of Mycobacterium tuberculosis, M. bovis, M. africanum and other related species. J. Gen. Microbiol. 1988, 134, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Sampson, S.L.; Warren, R.M.; Gey van Pittius, N.C.; Newton-Foot, M. Iron acquisition strategies in mycobacteria. Tuberculosis 2015, 95, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Gobin, J.; Moore, C.H.; Reeve, J.R., Jr.; Wong, D.K.; Gibson, B.W.; Horwitz, M.A. Iron acquisition by Mycobacterium tuberculosis: Isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. USA 1995, 92, 5189–5193. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Horwitz, M.A. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 1996, 184, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Farhana, A.; Kumar, S.; Rathore, S.S.; Ghosh, P.C.; Ehtesham, N.Z.; Tyagi, A.K.; Hasnain, S.E. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS ONE 2008, 3, e2087. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.; Pal, R.; Fatima, Z. Iron Acquisition Mechanisms: Promising Target Against Mycobacterium tuberculosis. Open Microbiol. J. 2015, 9, 91–97. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Wang, S.; Smith, I.; Rodriguez, G.M. The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J. Bacteriol. 2010, 192, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Smith, I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol. 2006, 188, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.M.; Jones, C.M.; Xi, Z.; Speer, A.; Danilchanka, O.; Doornbos, K.S.; Sun, P.; Wu, F.; Tian, C.; Niederweis, M. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003120. [Google Scholar] [CrossRef]
- Jones, C.M.; Wells, R.M.; Madduri, A.V.; Renfrow, M.B.; Ratledge, C.; Moody, D.B.; Niederweis, M. Self-poisoning of Mycobacterium tuberculosis by interrupting siderophore recycling. Proc. Natl. Acad. Sci. USA 2014, 111, 1945–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, L.E.; Sello, J.; Keating, T.A.; Weinreb, P.H.; Walsh, C.T. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998, 5, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Sritharan, M. Iron Homeostasis in Mycobacterium tuberculosis: Mechanistic Insights into Siderophore-Mediated Iron Uptake. J. Bacteriol. 2016, 198, 2399–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E., 3rd. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groschel, M.I.; Sayes, F.; Simeone, R.; Majlessi, L.; Brosch, R. ESX secretion systems: Mycobacterial evolution to counter host immunity. Nat. Rev. Microbiol. 2016, 14, 677–691. [Google Scholar] [CrossRef]
- Serafini, A.; Boldrin, F.; Palu, G.; Manganelli, R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: Essentiality and rescue by iron and zinc. J. Bacteriol. 2009, 191, 6340–6344. [Google Scholar] [CrossRef] [PubMed]
- Serafini, A.; Pisu, D.; Palu, G.; Rodriguez, G.M.; Manganelli, R. The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e78351. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, M.S.; Unnikrishnan, M.; McConnell, M.J.; Borowsky, M.; Cheng, T.Y.; Siddiqi, N.; Fortune, S.M.; Moody, D.B.; Rubin, E.J. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. USA 2009, 106, 18792–18797. [Google Scholar] [CrossRef] [Green Version]
- Tufariello, J.M.; Chapman, J.R.; Kerantzas, C.A.; Wong, K.W.; Vilcheze, C.; Jones, C.M.; Cole, L.E.; Tinaztepe, E.; Thompson, V.; Fenyo, D.; et al. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl. Acad. Sci. USA 2016, 113, E348–E357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.M.; Niederweis, M. Mycobacterium tuberculosis can utilize heme as an iron source. J. Bacteriol. 2011, 193, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Wren, B.W.; Stabler, R.A.; Das, S.S.; Butcher, P.D.; Mangan, J.A.; Clarke, J.D.; Casali, N.; Parish, T.; Stoker, N.G. Characterization of a haemolysin from Mycobacterium tuberculosis with homology to a virulence factor of Serpulina hyodysenteriae. Microbiology 1998, 144 Pt 5, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Tullius, M.V.; Harmston, C.A.; Owens, C.P.; Chim, N.; Morse, R.P.; McMath, L.M.; Iniguez, A.; Kimmey, J.M.; Sawaya, M.R.; Whitelegge, J.P.; et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc. Natl. Acad. Sci. USA 2011, 108, 5051–5056. [Google Scholar] [CrossRef]
- Owens, C.P.; Chim, N.; Graves, A.B.; Harmston, C.A.; Iniguez, A.; Contreras, H.; Liptak, M.D.; Goulding, C.W. The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J. Biol. Chem. 2013, 288, 21714–21728. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Speer, A.; Lin, K.; Ehrt, S.; Niederweis, M. PPE Surface Proteins Are Required for Heme Utilization by Mycobacterium tuberculosis. mBio. 2017, 8, e01720-16. [Google Scholar] [CrossRef] [PubMed]
- Tullius, M.V.; Nava, S.; Horwitz, M.A. PPE37 Is Essential for Mycobacterium tuberculosis Heme-Iron Acquisition (HIA), and a Defective PPE37 in Mycobacterium bovis BCG Prevents HIA. Infect. Immun. 2019, 87, e00540-18. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Farhana, A.; Pancsa, R.; Arora, S.K.; Srinivasan, A.; Tyagi, A.K.; Babu, M.M.; Ehtesham, N.Z.; Hasnain, S.E. Contrasting Function of Structured N-Terminal and Unstructured C-Terminal Segments of Mycobacterium tuberculosis PPE37 Protein. mBio 2018, 9, e01712-17. [Google Scholar] [CrossRef] [PubMed]
- Owens, C.P.; Chim, N.; Goulding, C.W. Insights on how the Mycobacterium tuberculosis heme uptake pathway can be used as a drug target. Future Med. Chem. 2013, 5, 1391–1403. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.V.; Puri, R.V.; Khera, A.; Tyagi, A.K. Iron storage proteins are essential for the survival and pathogenesis of Mycobacterium tuberculosis in THP-1 macrophages and the guinea pig model of infection. J. Bacteriol. 2012, 194, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Khare, G.; Nangpal, P.; Tyagi, A.K. Differential Roles of Iron Storage Proteins in Maintaining the Iron Homeostasis in Mycobacterium tuberculosis. PLoS ONE 2017, 12, e0169545. [Google Scholar] [CrossRef]
- Pandey, R.; Rodriguez, G.M. A ferritin mutant of Mycobacterium tuberculosis is highly susceptible to killing by antibiotics and is unable to establish a chronic infection in mice. Infect. Immun. 2012, 80, 3650–3659. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Voskuil, M.I.; Gold, B.; Schoolnik, G.K.; Smith, I. ideR, An essential gene in mycobacterium tuberculosis: Role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 2002, 70, 3371–3381. [Google Scholar] [CrossRef]
- Pandey, S.D.; Choudhury, M.; Yousuf, S.; Wheeler, P.R.; Gordon, S.V.; Ranjan, A.; Sritharan, M. Iron-regulated protein HupB of Mycobacterium tuberculosis positively regulates siderophore biosynthesis and is essential for growth in macrophages. J. Bacteriol. 2014, 196, 1853–1865. [Google Scholar] [CrossRef]
- Pandey, R.; Rodriguez, G.M. IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis. Mol. Microbiol. 2014, 91, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 4653. [Google Scholar] [CrossRef]
- Kumar, A.; Toledo, J.C.; Patel, R.P.; Lancaster, J.R., Jr.; Steyn, A.J. Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. USA 2007, 104, 11568–11573. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Crossman, D.K.; Mai, D.; Guidry, L.; Voskuil, M.I.; Renfrow, M.B.; Steyn, A.J. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 2009, 5, e1000545. [Google Scholar] [CrossRef]
- Geiman, D.E.; Raghunand, T.R.; Agarwal, N.; Bishai, W.R. Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob. Agents Chemother. 2006, 50, 2836–2841. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Guidry, L.; Narasimhulu, K.V.; Mai, D.; Trombley, J.; Redding, K.E.; Giles, G.I.; Lancaster, J.R., Jr.; Steyn, A.J. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. USA 2007, 104, 11562–11567. [Google Scholar] [CrossRef]
- Wu, J.; Ru, H.W.; Xiang, Z.H.; Jiang, J.; Wang, Y.C.; Zhang, L.; Liu, J. WhiB4 Regulates the PE/PPE Gene Family and is Essential for Virulence of Mycobacterium marinum. Sci. Rep. 2017, 7, 3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casonato, S.; Cervantes Sanchez, A.; Haruki, H.; Rengifo Gonzalez, M.; Provvedi, R.; Dainese, E.; Jaouen, T.; Gola, S.; Bini, E.; Vicente, M.; et al. WhiB5, a transcriptional regulator that contributes to Mycobacterium tuberculosis virulence and reactivation. Infect. Immun. 2012, 80, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Y.; Cumming, B.M.; Lu, P.; Feng, L.; Deng, J.; Steyn, A.J.; Chen, S. Mycobacterial WhiB6 Differentially Regulates ESX-1 and the Dos Regulon to Modulate Granuloma Formation and Virulence in Zebrafish. Cell Rep. 2016, 16, 2512–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.M.; Collins, D.M. ahpC, a gene involved in isoniazid resistance of the Mycobacterium tuberculosis complex. Mol. Microbiol. 1996, 19, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Piddington, D.L.; Fang, F.C.; Laessig, T.; Cooper, A.M.; Orme, I.M.; Buchmeier, N.A. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect. Immun. 2001, 69, 4980–4987. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Fan, Q.; Bao, L. The role of superoxide dismutase in the survival of Mycobacterium tuberculosis in macrophages. Jpn. J. Infect. Dis. 2013, 66, 480–488. [Google Scholar] [CrossRef]
- Edwards, K.M.; Cynamon, M.H.; Voladri, R.K.; Hager, C.C.; DeStefano, M.S.; Tham, K.T.; Lakey, D.L.; Bochan, M.R.; Kernodle, D.S. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 2001, 164, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef]
- Kumar, A.; Farhana, A.; Guidry, L.; Saini, V.; Hondalus, M.; Steyn, A.J. Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 2011, 13, e39. [Google Scholar] [CrossRef]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Byrd, T.F.; Horwitz, M.A. Interferon gamma-activated human monocytes downregulate transferrin receptors and inhibit the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. J. Clin. Investig. 1989, 83, 1457–1465. [Google Scholar] [CrossRef]
- Garcia-Montoya, I.A.; Cendon, T.S.; Arevalo-Gallegos, S.; Rascon-Cruz, Q. Lactoferrin a multiple bioactive protein: An overview. Biochim. Biophys. Acta 2012, 1820, 226–236. [Google Scholar] [CrossRef]
- Ward, P.P.; Paz, E.; Conneely, O.M. Multifunctional roles of lactoferrin: A critical overview. Cell. Mol. Life Sci. 2005, 62, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Silva-Gomes, S.; Bouton, C.; Silva, T.; Santambrogio, P.; Rodrigues, P.; Appelberg, R.; Gomes, M.S. Mycobacterium avium infection induces H-ferritin expression in mouse primary macrophages by activating Toll-like receptor 2. PLoS ONE 2013, 8, e82874. [Google Scholar] [CrossRef]
- Momotani, E.; Furugouri, K.; Obara, Y.; Miyata, Y.; Ishikawa, Y.; Yoshino, T. Immunohistochemical distribution of ferritin, lactoferrin, and transferrin in granulomas of bovine paratuberculosis. Infect. Immun. 1986, 52, 623–627. [Google Scholar]
- Reddy, V.P.; Chinta, K.C.; Saini, V.; Glasgow, J.N.; Hull, T.D.; Traylor, A.; Rey-Stolle, F.; Soares, M.P.; Madansein, R.; Rahman, M.A.; et al. Ferritin H Deficiency in Myeloid Compartments Dysregulates Host Energy Metabolism and Increases Susceptibility to Mycobacterium tuberculosis Infection. Front. Immunol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.; Skamene, E.; Forget, A. Genetic control of natural resistance to Mycobacterium bovis (BCG) in mice. J. Immunol. 1981, 127, 2417–2421. [Google Scholar]
- Skamene, E.; Gros, P.; Forget, A.; Patel, P.J.; Nesbitt, M.N. Regulation of resistance to leprosy by chromosome 1 locus in the mouse. Immunogenetics 1984, 19, 117–124. [Google Scholar] [CrossRef]
- Stach, J.L.; Gros, P.; Forget, A.; Skamene, E. Phenotypic expression of genetically-controlled natural resistance to Mycobacterium bovis (BCG). J. Immunol. 1984, 132, 888–892. [Google Scholar] [PubMed]
- Rodrigues, P.N.; Gomes, S.S.; Neves, J.V.; Gomes-Pereira, S.; Correia-Neves, M.; Nunes-Alves, C.; Stolte, J.; Sanchez, M.; Appelberg, R.; Muckenthaler, M.U.; et al. Mycobacteria-induced anaemia revisited: A molecular approach reveals the involvement of NRAMP1 and lipocalin-2, but not of hepcidin. Immunobiology 2011, 216, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, S.M.; Malo, D.; Vogan, K.; Skamene, E.; Gros, P. Natural resistance to infection with intracellular parasites: Isolation of a candidate for Bcg. Cell 1993, 73, 469–485. [Google Scholar] [CrossRef]
- Vidal, S.; Tremblay, M.L.; Govoni, G.; Gauthier, S.; Sebastiani, G.; Malo, D.; Skamene, E.; Olivier, M.; Jothy, S.; Gros, P. The Ity/Lsh/Bcg locus: Natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J. Exp. Med. 1995, 182, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.M.; Pinner, E.; Lepage, P.; Gauthier, S.; Gros, P. Natural resistance to intracellular infections: Nramp1 encodes a membrane phosphoglycoprotein absent in macrophages from susceptible (Nramp1 D169) mouse strains. J. Immunol. 1996, 157, 3559–3568. [Google Scholar]
- Wessling-Resnick, M. Nramp1 and Other Transporters Involved in Metal Withholding during Infection. J. Biol. Chem. 2015, 290, 18984–18990. [Google Scholar] [CrossRef] [PubMed]
- Stead, W.W.; Senner, J.W.; Reddick, W.T.; Lofgren, J.P. Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N. Engl. J. Med. 1990, 322, 422–427. [Google Scholar] [CrossRef]
- Hoal, E.G.; Lewis, L.A.; Jamieson, S.E.; Tanzer, F.; Rossouw, M.; Victor, T.; Hillerman, R.; Beyers, N.; Blackwell, J.M.; Van Helden, P.D. SLC11A1 (NRAMP1) but not SLC11A2 (NRAMP2) polymorphisms are associated with susceptibility to tuberculosis in a high-incidence community in South Africa. Int. J. Tuberc. Lung Dis. 2004, 8, 1464–1471. [Google Scholar]
- Fernandez-Mestre, M.; Villasmil, A.; Takiff, H.; Alcala, Z.F. NRAMP1 and VDR Gene Polymorphisms in Susceptibility to Tuberculosis in Venezuelan Population. Dis. Markers 2015, 2015, 860628. [Google Scholar] [CrossRef]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [Green Version]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [Green Version]
- Besson-Fournier, C.; Latour, C.; Kautz, L.; Bertrand, J.; Ganz, T.; Roth, M.P.; Coppin, H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012, 120, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Poli, M.; Girelli, D.; Campostrini, N.; Maccarinelli, F.; Finazzi, D.; Luscieti, S.; Nai, A.; Arosio, P. Heparin: A potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011, 117, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Brkljacic, J.; Pauk, M.; Erjavec, I.; Cipcic, A.; Grgurevic, L.; Zadro, R.; Inman, G.J.; Vukicevic, S. Exogenous heparin binds and inhibits bone morphogenetic protein 6 biological activity. Int. Orthop. 2013, 37, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Weiss, G. Iron metabolism in the anemia of chronic disease. Biochim. Biophys. Acta 2009, 1790, 682–693. [Google Scholar] [CrossRef]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef]
- Sow, F.B.; Nandakumar, S.; Velu, V.; Kellar, K.L.; Schlesinger, L.S.; Amara, R.R.; Lafuse, W.P.; Shinnick, T.M.; Sable, S.B. Mycobacterium tuberculosis components stimulate production of the antimicrobial peptide hepcidin. Tuberculosis 2011, 91, 314–321. [Google Scholar] [CrossRef]
- Harrington-Kandt, R.; Stylianou, E.; Eddowes, L.A.; Lim, P.J.; Stockdale, L.; Pinpathomrat, N.; Bull, N.; Pasricha, J.; Ulaszewska, M.; Beglov, Y.; et al. Hepcidin deficiency and iron deficiency do not alter tuberculosis susceptibility in a murine M.tb infection model. PLoS ONE 2018, 13, e0191038. [Google Scholar] [CrossRef]
- Agoro, R.; Benmerzoug, S.; Rose, S.; Bouyer, M.; Gozzelino, R.; Garcia, I.; Ryffel, B.; Quesniaux, V.F.J.; Mura, C. An Iron-Rich Diet Decreases the Mycobacterial Burden and Correlates With Hepcidin Upregulation, Lower Levels of Proinflammatory Mediators, and Increased T-Cell Recruitment in a Model of Mycobacterium bovis Bacille Calmette-Guerin Infection. J. Infect. Dis. 2017, 216, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.E.; Sandgren, A.; Cherayil, B.J.; Murray, M.; Wessling-Resnick, M. Role of ferroportin in macrophage-mediated immunity. Infect. Immun. 2010, 78, 5099–5106. [Google Scholar] [CrossRef]
- Baker, M.A.; Wilson, D.; Wallengren, K.; Sandgren, A.; Iartchouk, O.; Broodie, N.; Goonesekera, S.D.; Sabeti, P.C.; Murray, M.B. Polymorphisms in the gene that encodes the iron transport protein ferroportin 1 influence susceptibility to tuberculosis. J. Infect. Dis. 2012, 205, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Kochan, I.; Golden, C.A.; Bukovic, J.A. Mechanism of tuberculostasis in mammalian serum. II. Induction of serum tuberculostasis in guinea pigs. J. Bacteriol. 1969, 100, 64–70. [Google Scholar]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Krause, A.; Neitz, S.; Magert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.A.; Paulsene, W.; Jide, X.; Ratledge, C.; Strong, R.K. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 2005, 13, 29–41. [Google Scholar] [CrossRef]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef]
- Saiga, H.; Nishimura, J.; Kuwata, H.; Okuyama, M.; Matsumoto, S.; Sato, S.; Matsumoto, M.; Akira, S.; Yoshikai, Y.; Honda, K.; et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J. Immunol. 2008, 181, 8521–8527. [Google Scholar] [CrossRef] [PubMed]
- Guglani, L.; Gopal, R.; Rangel-Moreno, J.; Junecko, B.F.; Lin, Y.; Berger, T.; Mak, T.W.; Alcorn, J.F.; Randall, T.D.; Reinhart, T.A.; et al. Lipocalin 2 regulates inflammation during pulmonary mycobacterial infections. PLoS ONE 2012, 7, e50052. [Google Scholar] [CrossRef] [PubMed]
- Toyonaga, T.; Matsuura, M.; Mori, K.; Honzawa, Y.; Minami, N.; Yamada, S.; Kobayashi, T.; Hibi, T.; Nakase, H. Lipocalin 2 prevents intestinal inflammation by enhancing phagocytic bacterial clearance in macrophages. Sci. Rep. 2016, 6, 35014. [Google Scholar] [CrossRef] [Green Version]
- Cronje, L.; Edmondson, N.; Eisenach, K.D.; Bornman, L. Iron and iron chelating agents modulate Mycobacterium tuberculosis growth and monocyte-macrophage viability and effector functions. FEMS Immunol. Med. Microbiol. 2005, 45, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schaible, U.E.; Collins, H.L.; Priem, F.; Kaufmann, S.H. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J. Exp. Med. 2002, 196, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Raghu, B.; Sarma, G.R.; Venkatesan, P. Effect of iron on the growth and siderophore production of mycobacteria. Biochem. Mol. Biol. Int. 1993, 31, 341–348. [Google Scholar]
- Rook, G.A.; Steele, J.; Ainsworth, M.; Champion, B.R. Activation of macrophages to inhibit proliferation of Mycobacterium tuberculosis: Comparison of the effects of recombinant gamma-interferon on human monocytes and murine peritoneal macrophages. Immunology 1986, 59, 333–338. [Google Scholar] [PubMed]
- Kochan, I.; Pellis, N.R.; Golden, C.A. Mechanism of Tuberculostasis in Mammalian Serum III. Neutralization of Serum Tuberculostasis by Mycobactin. Infect. Immun. 1971, 3, 553–558. [Google Scholar] [PubMed]
- Gomes, M.S.; Dom, G.; Pedrosa, J.; Boelaert, J.R.; Appelberg, R. Effects of iron deprivation on Mycobacterium avium growth. Tuber Lung Dis. 1999, 79, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Rajiv, J.; Dam, T.; Kumar, S.; Bose, M.; Aggarwal, K.K.; Babu, C.R. Inhibition of the in-vitro growth of Mycobacterium tuberculosis by a phytosiderophore. J. Med. Microbiol. 2001, 50, 916–918. [Google Scholar] [CrossRef] [Green Version]
- Lamb, A.L. Breaking a pathogen’s iron will: Inhibiting siderophore production as an antimicrobial strategy. Biochim. Biophys. Acta 2015, 1854, 1054–1070. [Google Scholar] [CrossRef]
- Silva-Gomes, S.; Appelberg, R.; Larsen, R.; Soares, M.P.; Gomes, M.S. Heme catabolism by heme oxygenase-1 confers host resistance to Mycobacterium infection. Infect. Immun. 2013, 81, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.; Buddle, B.M. Iron modulates the replication of virulent Mycobacterium bovis in resting and activated bovine and possum macrophages. Vet. Immunol. Immunopathol. 2005, 107, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.S.; Boelaert, J.R.; Appelberg, R. Role of iron in experimental Mycobacterium avium infection. J. Clin. Virol. 2001, 20, 117–122. [Google Scholar] [CrossRef]
- Wheeler, W.C.; Hanks, J.H. Utilization of External Growth Factors by Intracellular Microbes: Mycobacterium Paratuberculosis and Wood Pigeon Mycobacteria. J. Bacteriol. 1965, 89, 889–896. [Google Scholar] [PubMed]
- Douvas, G.S.; May, M.H.; Crowle, A.J. Transferrin, Iron, and Serum-Lipids Enhance or Inhibit Mycobacterium-Avium Replication in Human Macrophages. J. Infect. Dis. 1993, 167, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [PubMed]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [Green Version]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Werner, E.R.; Werner-Felmayer, G.; Wachter, H. Iron modulates interferon-gamma effects in the human myelomonocytic cell line THP-1. Exp. Hematol. 1992, 20, 605–610. [Google Scholar]
- Byrd, T.F. Tumor necrosis factor alpha (TNFalpha) promotes growth of virulent Mycobacterium tuberculosis in human monocytes iron-mediated growth suppression is correlated with decreased release of TNFalpha from iron-treated infected monocytes. J. Clin. Investig. 1997, 99, 2518–2529. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Khoshdel, Z.; Amirghofran, Z. The effect of an iron (III) chelator, silybin, on the proliferation and cell cycle of Jurkat cells: A comparison with desferrioxamine. Eur. J. Pharmacol. 2008, 589, 1–7. [Google Scholar] [CrossRef]
- Lounis, N.; Truffot-Pernot, C.; Grosset, J.; Gordeuk, V.R.; Boelaert, J.R. Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 2001, 20, 123–126. [Google Scholar] [CrossRef]
- Stefanova, D.; Raychev, A.; Arezes, J.; Ruchala, P.; Gabayan, V.; Skurnik, M.; Dillon, B.J.; Horwitz, M.A.; Ganz, T.; Bulut, Y.; et al. Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood 2017, 130, 245–257. [Google Scholar] [CrossRef]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairo, G.; Recalcati, S.; Montosi, G.; Castrusini, E.; Conte, D.; Pietrangelo, A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood 1997, 89, 2546–2553. [Google Scholar] [PubMed]
- Gomes-Pereira, S.; Rodrigues, P.N.; Appelberg, R.; Gomes, M.S. Increased susceptibility to Mycobacterium avium in hemochromatosis protein HFE-deficient mice. Infect. Immun. 2008, 76, 4713–4719. [Google Scholar] [CrossRef]
- Rolph, M.S.; Raupach, B.; Kobernick, H.H.; Collins, H.L.; Perarnau, B.; Lemonnier, F.A.; Kaufmann, S.H. MHC class Ia-restricted T cells partially account for beta2-microglobulin-dependent resistance to Mycobacterium tuberculosis. Eur. J. Immunol. 2001, 31, 1944–1949. [Google Scholar] [CrossRef]
- Kasvosve, I.; Gangaidzo, I.T.; Gomo, Z.A.; Gordeuk, V.R. African iron overload. Acta Clin. Belg. 2000, 55, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R. African iron overload. Semin. Hematol. 2002, 39, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; McLaren, C.E.; MacPhail, A.P.; Deichsel, G.; Bothwell, T.H. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan’s 1929 thesis revisited. Blood 1996, 87, 3470–3476. [Google Scholar]
- Gangaidzo, I.T.; Moyo, V.M.; Mvundura, E.; Aggrey, G.; Murphree, N.L.; Khumalo, H.; Saungweme, T.; Kasvosve, I.; Gomo, Z.A.; Rouault, T.; et al. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 2001, 184, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Minchella, P.A.; Armitage, A.E.; Darboe, B.; Jallow, M.W.; Drakesmith, H.; Jaye, A.; Prentice, A.M.; McDermid, J.M. Elevated hepcidin at HIV diagnosis is associated with incident tuberculosis in a retrospective cohort study. Int. J. Tuberc. Lung Dis. 2014, 18, 1337–1339. [Google Scholar] [CrossRef]
- McDermid, J.M.; Hennig, B.J.; van der Sande, M.; Hill, A.V.; Whittle, H.C.; Jaye, A.; Prentice, A.M. Host iron redistribution as a risk factor for incident tuberculosis in HIV infection: An 11-year retrospective cohort study. BMC Infect. Dis. 2013, 13, 48. [Google Scholar] [CrossRef]
- Das, B.S.; Devi, U.; Mohan Rao, C.; Srivastava, V.K.; Rath, P.K.; Das, B.S. Effect of iron supplementation on mild to moderate anaemia in pulmonary tuberculosis. Br. J. Nutr. 2003, 90, 541–550. [Google Scholar] [Green Version]
- Isanaka, S.; Aboud, S.; Mugusi, F.; Bosch, R.J.; Willett, W.C.; Spiegelman, D.; Duggan, C.; Fawzi, W.W. Iron status predicts treatment failure and mortality in tuberculosis patients: A prospective cohort study from Dar es Salaam, Tanzania. PLoS ONE 2012, 7, e37350. [Google Scholar] [CrossRef]
- Chandrasekaran, P.; Saravanan, N.; Bethunaickan, R.; Tripathy, S. Malnutrition: Modulator of Immune Responses in Tuberculosis. Front. Immunol. 2017, 8, 1316. [Google Scholar] [CrossRef] [Green Version]
- Chandra, R.K.; Saraya, A.K. Impaired immunocompetence associated with iron deficiency. J. Pediatr. 1975, 86, 899–902. [Google Scholar] [CrossRef]
- Joynson, D.H.; Walker, D.M.; Jacobs, A.; Dolby, A.E. Defect of cell-mediated immunity in patients with iron-deficiency anaemia. Lancet 1972, 2, 1058–1059. [Google Scholar] [CrossRef]
- Oppenheimer, S.J. Iron and its relation to immunity and infectious disease. J. Nutr. 2001, 131, 616S–633S. [Google Scholar] [CrossRef]
- Esan, M.O.; van Hensbroek, M.B.; Nkhoma, E.; Musicha, C.; White, S.A.; Ter Kuile, F.O.; Phiri, K.S. Iron supplementation in HIV-infected Malawian children with anemia: A double-blind, randomized, controlled trial. Clin. Infect. Dis. 2013, 57, 1626–1634. [Google Scholar] [CrossRef]
- Sipahi, T.; Akar, N.; Egin, Y.; Cin, S. Serum interleukin-2 and interleukin-6 levels in iron deficiency anemia. Pediatr. Hematol. Oncol. 1998, 15, 69–73. [Google Scholar] [CrossRef]
- Rodriguez, L.; Gonzalez, C.; Flores, L.; Jimenez-Zamudio, L.; Graniel, J.; Ortiz, R. Assessment by flow cytometry of cytokine production in malnourished children. Clin. Diagn. Lab. Immunol. 2005, 12, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Hassan, T.H.; Badr, M.A.; Karam, N.A.; Zkaria, M.; El Saadany, H.F.; Abdel Rahman, D.M.; Shahbah, D.A.; Al Morshedy, S.M.; Fathy, M.; Esh, A.M.; et al. Impact of iron deficiency anemia on the function of the immune system in children. Medicine 2016, 95, e5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef]
- Deschemin, J.C.; Noordine, M.L.; Remot, A.; Willemetz, A.; Afif, C.; Canonne-Hergaux, F.; Langella, P.; Karim, Z.; Vaulont, S.; Thomas, M.; et al. The microbiota shifts the iron sensing of intestinal cells. FASEB J. 2016, 30, 252–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agoro, R.; Mura, C. Iron Supplementation Therapy, A Friend and Foe of Mycobacterial Infections? Pharmaceuticals 2019, 12, 75. https://doi.org/10.3390/ph12020075
Agoro R, Mura C. Iron Supplementation Therapy, A Friend and Foe of Mycobacterial Infections? Pharmaceuticals. 2019; 12(2):75. https://doi.org/10.3390/ph12020075
Chicago/Turabian StyleAgoro, Rafiou, and Catherine Mura. 2019. "Iron Supplementation Therapy, A Friend and Foe of Mycobacterial Infections?" Pharmaceuticals 12, no. 2: 75. https://doi.org/10.3390/ph12020075