Retinal Gene Distribution and Functionality Implicated in Inherited Retinal Degenerations Can Reveal Disease-Relevant Pathways for Pharmacologic Intervention


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Tissue Isolation
2.2. RNA Isolation
2.3. Preparation of RNA-Seq Library
2.4. RNA-Seq, Read Mappin, g and Determination of Reads per Kilobase per Million Reads (RPKM)
2.5. Pathway Analysis
2.6. Statistical Analyses
3. Results
3.1. Geographic Expression Profile of Known Retinal Disease Genes in the Primate Retina
3.2. Pathway and Overrepresentation Analysis of RetNet Disease Genes
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rattner, A.; Sun, H.; Nathans, J. Molecular genetics of human retinal disease. Annu. Rev. Genet. 1999, 33, 89–131. [Google Scholar] [CrossRef]
- Bessant, D.A.; Ali, R.R.; Bhattacharya, S.S. Molecular genetics and prospects for therapy of the inherited retinal dystrophies. Curr. Opin. Genet. Dev. 2001, 11, 307–316. [Google Scholar] [CrossRef]
- Daiger, S.P.; Rossiter, B.J.F.; Greenberg, J.; Christoffels, A.; Hide, W. Data services and software for identifying genes and mutations causing retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1998, 39, S295. [Google Scholar]
- Sorrentino, F.S.; Gallenga, C.E.; Bonifazzi, C.; Perri, P. A challenge to the striking genotypic heterogeneity of retinitis pigmentosa: A better understanding of the pathophysiology using the newest genetic strategies. Eye (Lond.) 2016, 30, 1542–1548. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Tuan, H.F.; Nguyen, D.H.; Sun, V.; Keser, V.; Bowne, S.J.; Sullivan, L.S.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 2014, 133, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schafer, E.; Roux, A.F.; Dafinger, C.; Bernd, A.; et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Investig. 2010, 120, 1812–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.F.; Chakarova, C.F.; Abd El-Aziz, M.M.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.H.; Chuang, J.Z. The cell biology of vision. J. Cell Biol. 2010, 190, 953–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A.; Sloan, K.R.; Kalina, R.E.; Hendrickson, A.E. Human photoreceptor topography. J. Comp. Neurol. 1990, 292, 497–523. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Brown, J., Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology 1998, 105, 1069–1075. [Google Scholar] [CrossRef]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The cone dysfunction syndromes. Br. J. Ophthalmol. 2004, 88, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.R.; Shekhar, K.; Yan, W.; Herrmann, D.; Sappington, A.; Bryman, G.S.; van Zyl, T.; Do, M.T.H.; Regev, A.; Sanes, J.R. Molecular Classification and Comparative Taxonomics of Foveal and Peripheral Cells in Primate Retina. Cell 2019, 176, 1222–1237. [Google Scholar] [CrossRef]
- Mustafi, D.; Kevany, B.M.; Bai, X.; Golczak, M.; Adams, M.D.; Wynshaw-Boris, A.; Palczewski, K. Transcriptome analysis reveals rod/cone photoreceptor specific signatures across mammalian retinas. Hum. Mol. Genet. 2016, 25, 4376–4388. [Google Scholar] [CrossRef]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Kellis, M.; Wold, B.; Snyder, M.P.; Bernstein, B.E.; Kundaje, A.; Marinov, G.K.; Ward, L.D.; Birney, E.; Crawford, G.E.; Dekker, J.; et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 2014, 111, 6131–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G. Human genetics. GTEx detects genetic effects. Science 2015, 348, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Sui, R.; Sallum, J.; van den Born, L.I.; Ajlan, R.; Khan, A.; den Hollander, A.I.; Cremers, F.P.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520. [Google Scholar] [CrossRef]
- Lee, H.; Lotery, A. Gene therapy for RPE65-mediated inherited retinal dystrophy completes phase 3. Lancet 2017, 390, 823–824. [Google Scholar] [CrossRef]
- Mustafi, D.; Kevany, B.M.; Genoud, C.; Okano, K.; Cideciyan, A.V.; Sumaroka, A.; Roman, A.J.; Jacobson, S.G.; Engel, A.; Adams, M.D.; et al. Defective photoreceptor phagocytosis in a mouse model of enhanced S-cone syndrome causes progressive retinal degeneration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3157–3176. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar]
- Davidson, A.E.; Sergouniotis, P.I.; Mackay, D.S.; Wright, G.A.; Waseem, N.H.; Michaelides, M.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Plagnol, V.; et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum. Mutat. 2013, 34, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, S.; Huang, L.; Yang, Y.; Zhang, L.; Yang, M.; Liu, W.; Ramasamy, K.; Jiang, Z.; Sundaresan, P.; et al. A splicing mutation in aryl hydrocarbon receptor associated with retinitis pigmentosa. Hum. Mol. Genet. 2018, 27, 2563–2572. [Google Scholar] [CrossRef]
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [Green Version]
- Palczewski, K. Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 2010, 31, 284–295. [Google Scholar] [CrossRef]
- Xu, M.; Eblimit, A.; Wang, J.; Li, J.; Wang, F.; Zhao, L.; Wang, X.; Xiao, N.; Li, Y.; Wong, L.J.; et al. ADIPOR1 Is Mutated in Syndromic Retinitis Pigmentosa. Hum. Mutat. 2016, 37, 246–249. [Google Scholar] [CrossRef]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T.; et al. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Mustafi, D.; Kevany, B.M.; Bai, X.; Maeda, T.; Sears, J.E.; Khalil, A.M.; Palczewski, K. Evolutionarily conserved long intergenic non-coding RNAs in the eye. Hum. Mol. Genet. 2013, 22, 2992–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Non-coding RNAome of RPE cells under oxidative stress suggests unknown regulative aspects of Retinitis pigmentosa etiopathogenesis. Sci. Rep. 2018, 8, 16638. [Google Scholar] [CrossRef]
- Chen, Y.; Palczewska, G.; Mustafi, D.; Golczak, M.; Dong, Z.; Sawada, O.; Maeda, T.; Maeda, A.; Palczewski, K. Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J. Clin. Investig. 2013, 123, 5119–5134. [Google Scholar] [CrossRef]
- Orban, T.; Leinonen, H.; Getter, T.; Dong, Z.; Sun, W.; Gao, S.; Veenstra, A.; Heidari-Torkabadi, H.; Kern, T.S.; Kiser, P.D.; et al. A Combination of G Protein-Coupled Receptor Modulators Protects Photoreceptors from Degeneration. J. Pharmacol. Exp. Ther. 2018, 364, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.M.; Fufa, T.D.; Bharti, K.; Brooks, B.P.; Hufnagel, R.B.; McGaughey, D.M. Identifying core biological processes distinguishing human eye tissues with precise systems-level gene expression analyses and weighted correlation networks. Hum. Mol. Genet. 2018, 27, 3325–3339. [Google Scholar] [CrossRef] [PubMed]
- Foltz, L.P.; Clegg, D.O. Patient-derived induced pluripotent stem cells for modelling genetic retinal dystrophies. Prog. Retin. Eye Res. 2019, 68, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Central | Peripheral | Pan-Retinal | Total |
|---|---|---|---|---|
| AMD | 1 | 7 | 3 | 11 |
| BBS | 1 | 11 | 8 | 20 |
| CSNB | 1 | 5 | 8 | 14 |
| LCA | 0 | 11 | 11 | 22 |
| MD | 2 | 5 | 4 | 11 |
| Optic Atrophy | 1 | 1 | 3 | 5 |
| Refsum Disease | 0 | 2 | 1 | 3 |
| RP | 4 | 33 | 38 | 75 |
| Usher Syndrome | 1 | 6 | 3 | 10 |
| Stickler Syndrome | 0 | 3 | 0 | 3 |
| Joubert Syndrome | 0 | 3 | 4 | 7 |
| Senior–Loken Syndrome | 0 | 3 | 2 | 5 |
| Other | 2 | 9 | 13 | 24 |
| Disease | Central | Peripheral | Pan-Retinal | Total |
|---|---|---|---|---|
| RP | 4 | 33 | 38 | 75 |
| RP only | 4 | 18 | 21 | 43 |
| RP, BBS | 0 | 1 | 2 | 3 |
| RP, CRD, CA | 0 | 0 | 1 | 1 |
| RP, MD | 0 | 1 | 3 | 4 |
| RP, Refsum | 0 | 2 | 1 | 3 |
| RP, LCA | 0 | 6 | 3 | 9 |
| RP, ESCS | 0 | 1 | 0 | 1 |
| RP, BBS, CRD | 0 | 0 | 1 | 1 |
| RP, CRD | 0 | 1 | 1 | 2 |
| RP, AMD, CRD | 0 | 2 | 0 | 2 |
| RP, CRD, LCA, MD | 0 | 0 | 1 | 1 |
| RP, CSNB | 0 | 1 | 2 | 3 |
| RP, Usher | 0 | 2 | 0 | 2 |
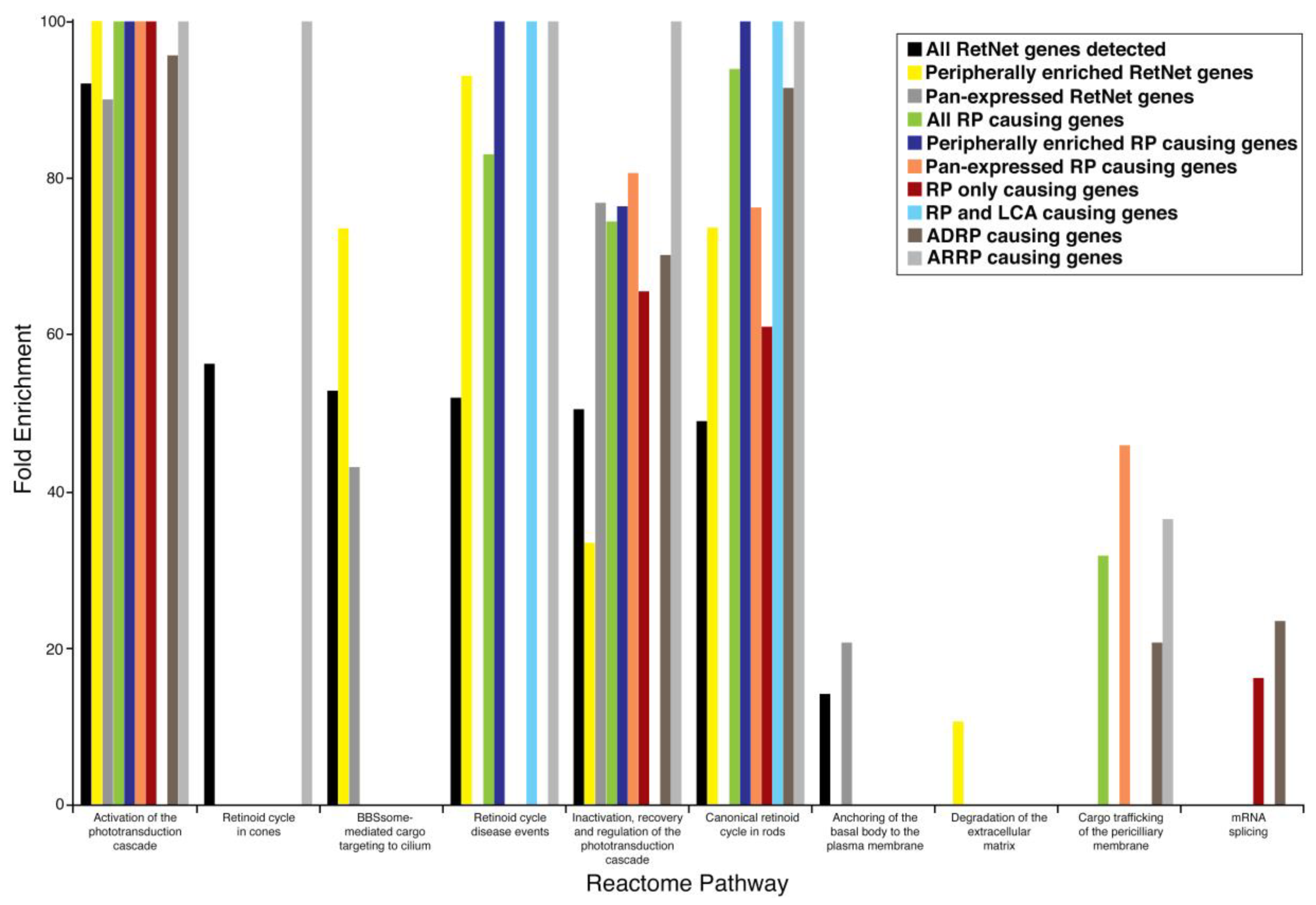
| Reactome Pathway | Fold Enrichment | p Value |
|---|---|---|
| All expressed RetNet genes (186) | ||
| Activation of phototransduction cascade | 92.07 | <0.00001 |
| The retinoid cycle in cones | 56.26 | 0.04240 |
| BBSome-mediated cargo targeting to cilium | 52.82 | <0.00001 |
| Retinoid cycle disease events | 51.93 | <0.00001 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 50.44 | <0.00001 |
| The canonical retinoid cycle in rods | 48.92 | <0.00001 |
| Anchoring of the basal body to the plasma membrane | 14.07 | <0.00001 |
| Peripherally enriched RetNet genes (85) | ||
| Activation of phototransduction cascade | >100 | <0.00001 |
| Retinoid cycle disease events | 93.02 | <0.00001 |
| BBSome-mediated cargo targeting to cilium | 73.61 | <0.00001 |
| The canonical retinoid cycle in rods | 73.61 | <0.00001 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 33.36 | 0.01300 |
| Degradation of the extracellular matrix | 10.59 | 0.04350 |
| Pan-expressed RetNet genes (86) | ||
| Activation of phototransduction cascade | 90.02 | 0.00026 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 76.83 | <0.00001 |
| BBSome-mediated cargo targeting to cilium | 43.05 | 0.00478 |
| Anchoring of the basal body to the plasma membrane | 20.63 | 0.00001 |
| Reactome Pathway | Fold Enrichment | p Value |
|---|---|---|
| All RP causing genes (75) | ||
| Activation of phototransduction cascade | >100 | <0.00001 |
| The canonical retinoid cycle in rods | 93.93 | <0.00001 |
| Retinoid cycle disease events | 83.01 | 0.00036 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 74.42 | <0.00001 |
| Cargo trafficking of the periciliary membrane | 31.74 | 0.00008 |
| Peripherally enriched RP genes (33) | ||
| Activation of phototransduction cascade | >100 | <0.00001 |
| Retinoid cycle disease events | >100 | 0.00002 |
| The canonical retinoid cycle in rods | >100 | <0.00001 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 76.38 | 0.00046 |
| Pan-expressed RP genes (38) | ||
| Activation of phototransduction cascade | >100 | 0.00179 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 80.62 | 0.00036 |
| The canonical retinoid cycle in rods | 76.24 | 0.01610 |
| Cargo trafficking of the periciliary membrane | 45.84 | 0.00339 |
| Genes causing only RP (43) | ||
| Activation of phototransduction cascade | >100 | 0.00002 |
| Inactivation, recovery, and regulation of the phototransduction cascade | 64.50 | 0.00091 |
| The canonical retinoid cycle in rods | 60.99 | 0.03180 |
| mRNA splicing | 16.12 | 0.00350 |
| Overlapping genes causing RP and LCA (9) | ||
| Retinoid cycle disease events | >100 | 0.02430 |
| The canonical retinoid cycle in rods | >100 | 0.00019 |
| Reactome Pathway | Fold Enrichment in ADRP (p Value) | Fold Enrichment in ARRP (p Value) |
|---|---|---|
| mRNA Splicing | 23.38 (0.00002) | - |
| Activation of phototransduction cascade | 95.65 (0.0113) | >100 (<0.00001) |
| Retinoid cycle disease events | - | >100 (<0.00001) |
| The canonical retinoid cycle in rods | 91.49 (0.00025) | >100 (<0.00001) |
| The retinoid cycle in cones | - | >100 (0.00009) |
| Inactivation, recovery, and regulation of the phototransduction cascade | 70.14 (0.00042) | >100 (<0.00001) |
| Cargo trafficking to the periciliary membrane | 20.63 (0.0483) | 36.4 (0.00009) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafi, D.; Arbabi, A.; Ameri, H.; Palczewski, K. Retinal Gene Distribution and Functionality Implicated in Inherited Retinal Degenerations Can Reveal Disease-Relevant Pathways for Pharmacologic Intervention. Pharmaceuticals 2019, 12, 74. https://doi.org/10.3390/ph12020074
Mustafi D, Arbabi A, Ameri H, Palczewski K. Retinal Gene Distribution and Functionality Implicated in Inherited Retinal Degenerations Can Reveal Disease-Relevant Pathways for Pharmacologic Intervention. Pharmaceuticals. 2019; 12(2):74. https://doi.org/10.3390/ph12020074
Chicago/Turabian StyleMustafi, Debarshi, Amirmohsen Arbabi, Hossein Ameri, and Krzysztof Palczewski. 2019. "Retinal Gene Distribution and Functionality Implicated in Inherited Retinal Degenerations Can Reveal Disease-Relevant Pathways for Pharmacologic Intervention" Pharmaceuticals 12, no. 2: 74. https://doi.org/10.3390/ph12020074