Determination of Picomolar Concentrations of Paraoxon in Human Urine by Fluorescence-Based Enzymatic Assay

 , , ,
, , ,  and
and
Abstract
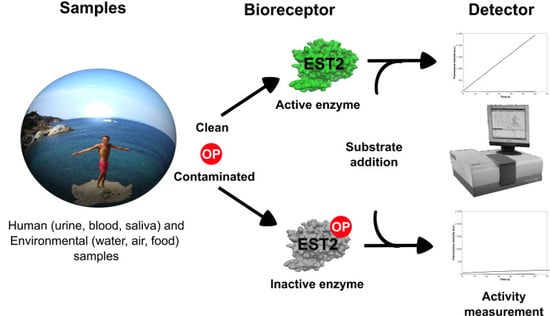
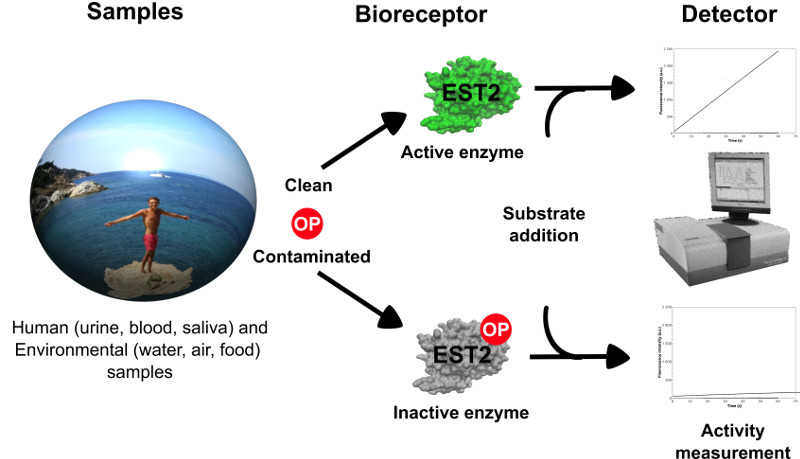
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Collection of Human Urine Samples
2.3. Enzyme Purification
2.4. MU Standard Calibration Curve in HEPES
2.5. Fluorescence Standard Enzymatic Assay
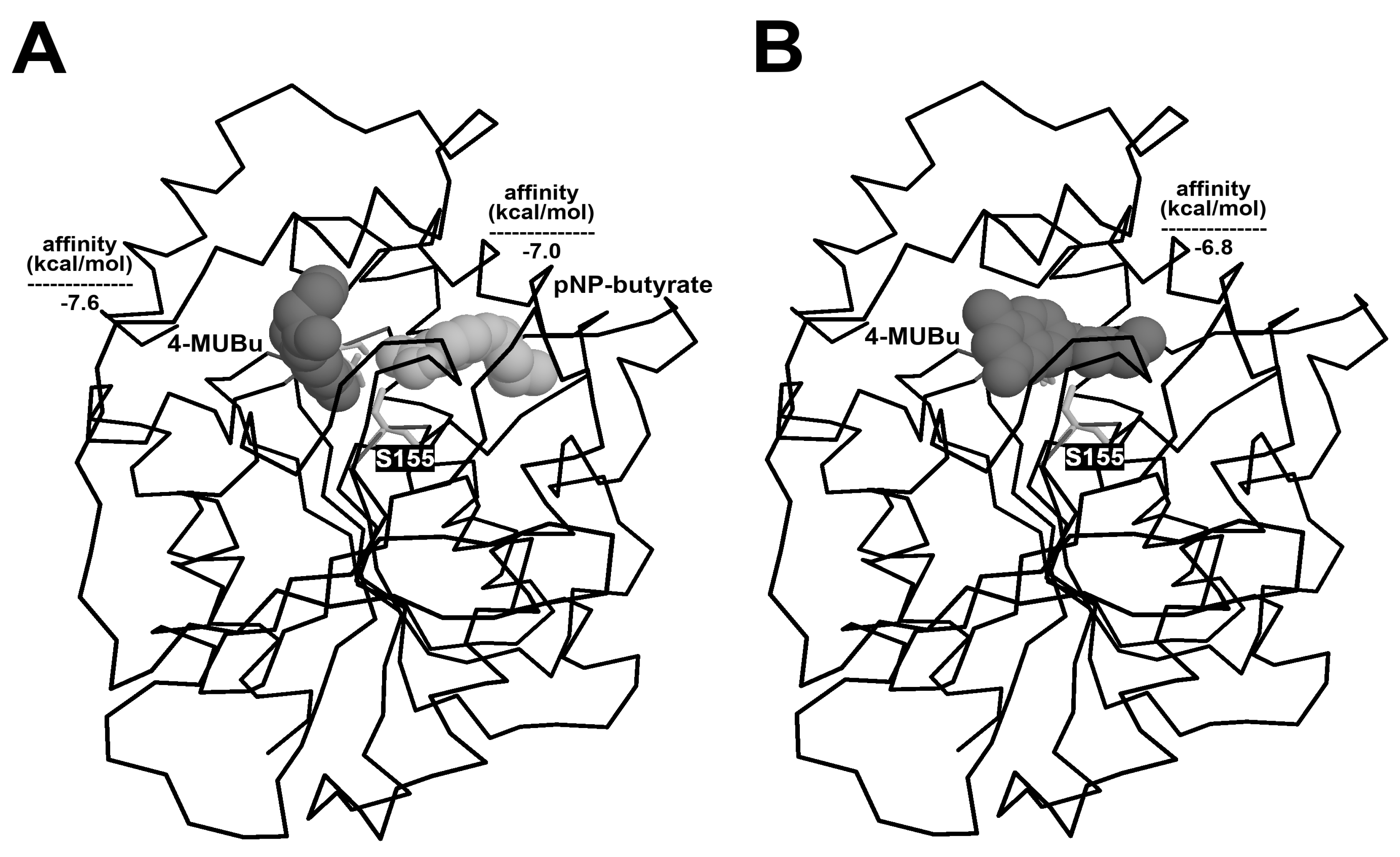
2.6. Docking Analysis
2.7. Kinetic Constants
2.8. Inhibition Assay of EST2 in Presence of Paraoxon
2.9. EST2 Residual Activity in Human Urine
2.10. Paraoxon Inhibition Assay of EST2 in Human Urine
3. Results and Discussion
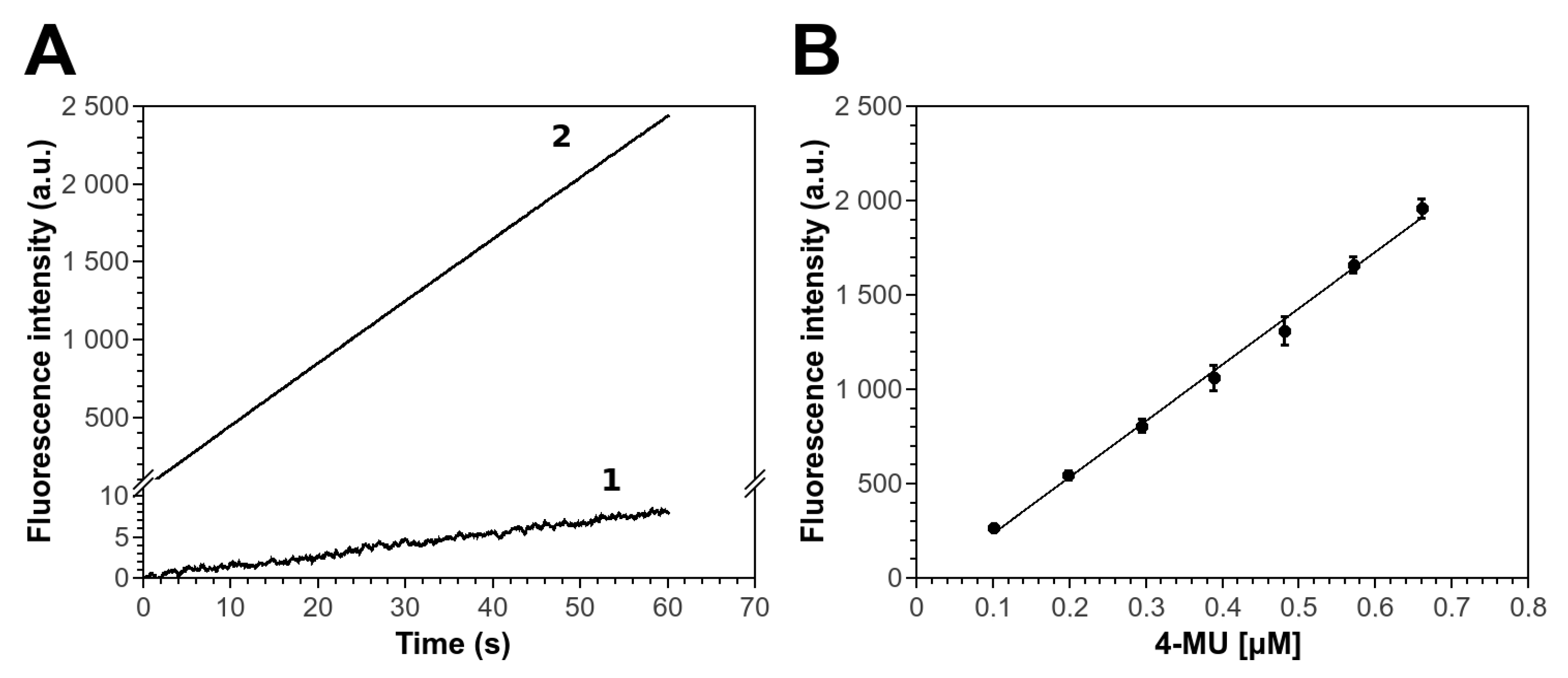
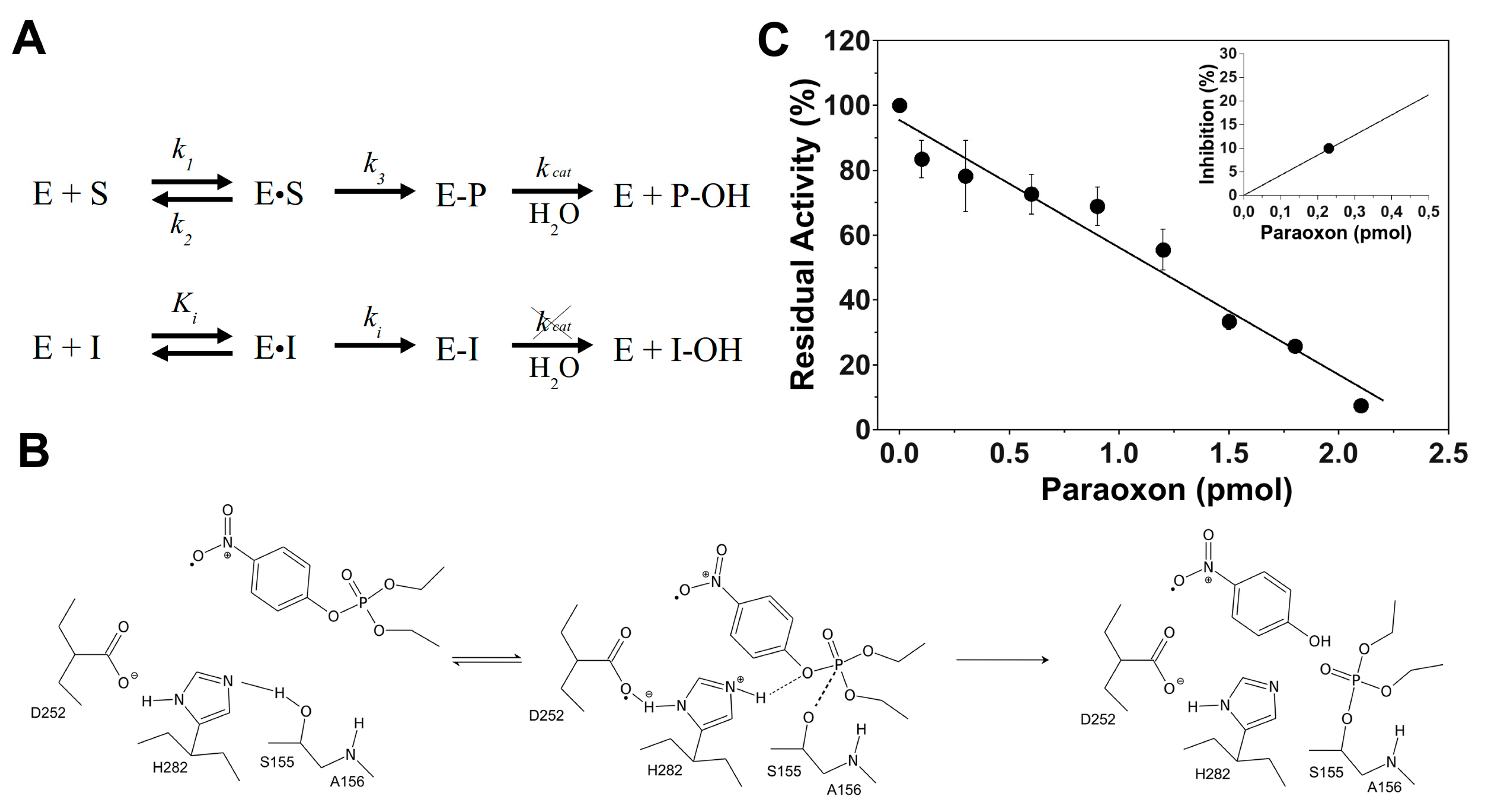
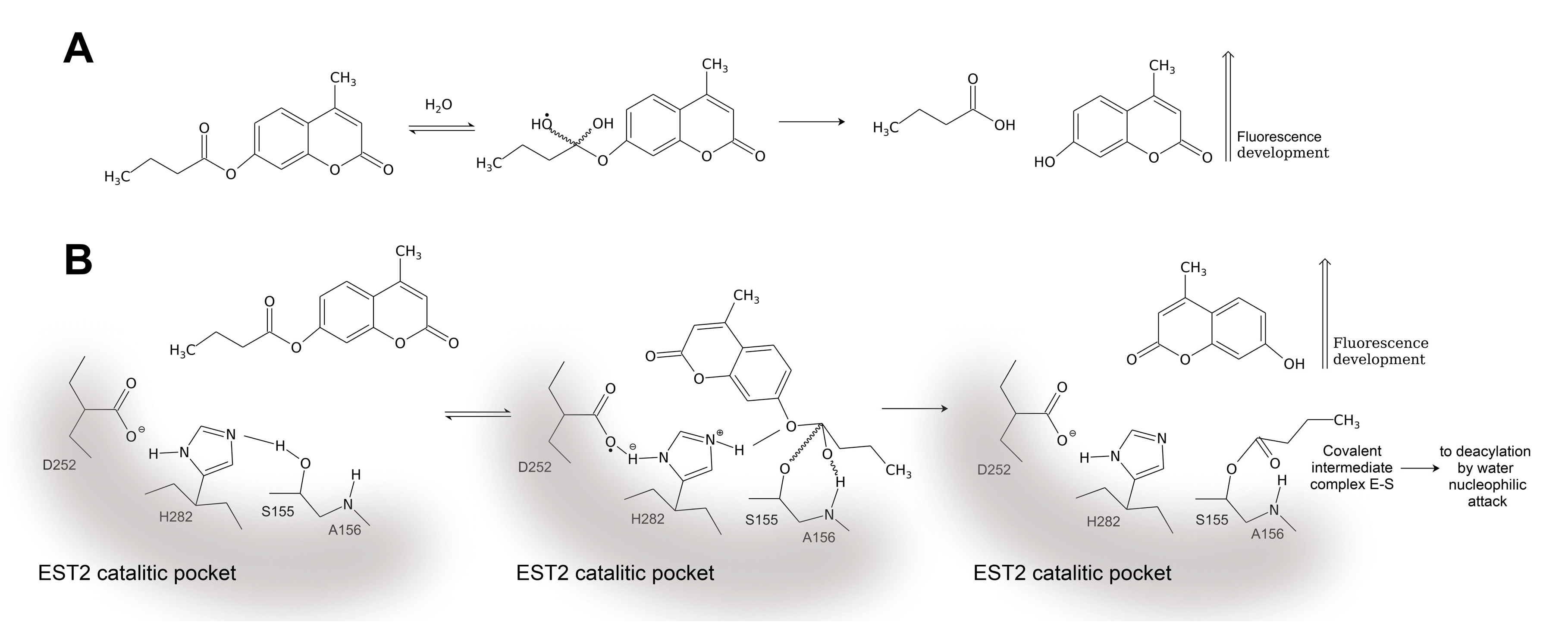
3.1. 4-MUBu Fluorescence Measurements and the Determination of Kinetics Parameters
3.2. EST2 Inhibition with Paraoxon Determined by Fluorescence Assay
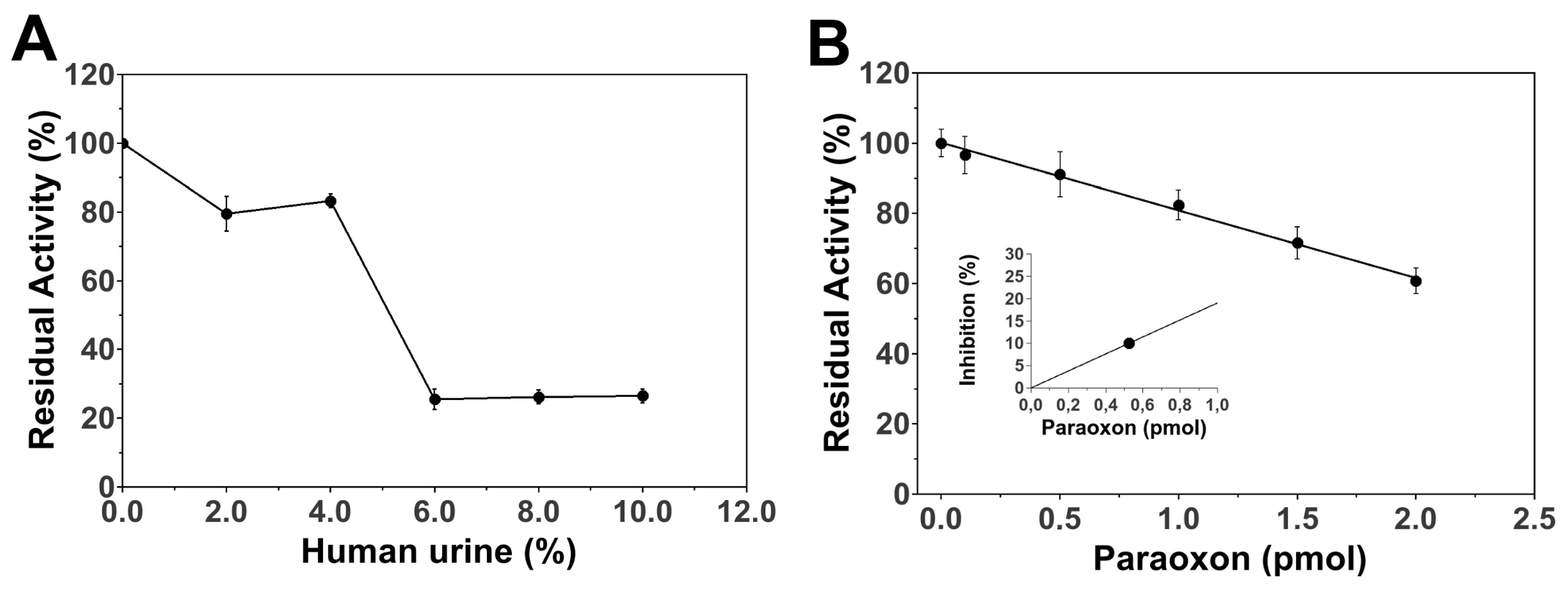
3.3. Paraoxon Determination in Human Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
- Collection of Human Blood Samples
- EST2 Residual Activity in Human Blood
- Inhibition Assay of EST2 in Human Blood
References
- Judson, R.; Richard, A.; Dix, D.J.; Houck, K.; Martin, M.; Kavlock, R.; Dellarco, V.; Henry, T.; Holderman, T.; Sayre, P.; et al. The toxicity data landscape for environmental chemicals. Environ. Health Perspect. 2009, 117, 685–695. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Goldman, L.R. Chemical safety, health care costs and the Affordable Care Act. Am. J. Ind. Med. 2014, 57, 1–3. [Google Scholar] [CrossRef]
- Bjørling-Poulsen, M.; Andersen, H.; Grandjean, P. Potential developmental neurotoxicity of pesticides used in Europe. Environ. Health 2008, 7, 50. [Google Scholar] [CrossRef]
- London, L.; Beseler, C.; Bouchard, M.F.; Bellinger, D.C.; Colosio, C.; Grandjean, P.; Harari, R.; Kootbodien, T.; Kromhout, H.; Little, F.; et al. Neurobehavioral and neurodevelopmental effects of pesticide exposures. Neurotoxicology 2012, 33, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Beard, J.D.; Hoppin, J.A.; Richards, M.; Alavanja, M.C.R.; Blair, A.; Sandler, D.P.; Kamel, F. Pesticide exposure and self-reported incident depression among wives in the Agricultural Health Study. Environ. Res. 2013, 126, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.A.; Wilkinson, S.C.; Blain, P.G.; Dunn, M.; Aust, G.A.; Williams, F.M. Percutaneous absorption and distribution of organophosphates (chlorpyrifos and dichlorvos) following dermal exposure and decontamination scenarios using in vitro human skin model. Toxicol. Lett. 2014, 229, 66–72. [Google Scholar] [CrossRef]
- Furlong, M.A.; Engel, S.M.; Barr, D.B.; Wolff, M.S. Prenatal exposure to organophosphate pesticides and reciprocal social behavior in childhood. Environ. Int. 2014, 70, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Rauh, V.A.; Garfinkel, R.; Perera, F.P.; Andrews, H.F.; Hoepner, L.; Barr, D.B.; Whitehead, R.; Tang, D.; Whyatt, R.W. Impact of prenatal chlorpyrifos exposure on neurodevelopment in the first 3 years of life among inner-city children. Pediatrics 2006, 118, e1845–e1859. [Google Scholar] [CrossRef]
- Rauh, V.; Arunajadai, S.; Horton, M.; Perera, F.; Hoepner, L.; Barr, D.B.; Whyatt, R. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ. Health Perspect. 2011, 119, 1196–1201. [Google Scholar] [CrossRef]
- Febbraio, F. Biochemical strategies for the detection and detoxification of toxic chemicals in the environment. World J. Biol. Chem. 2017, 8, 13–20. [Google Scholar] [CrossRef]
- Gervais, G.; Brosillon, S.; Laplanche, A.; Helen, C. Ultra-pressure liquid chromatography-electrospray tandem mass spectrometry for multiresidue determination of pesticides in water. J. Chromatogr. A 2008, 1202, 163–172. [Google Scholar] [CrossRef]
- Hengel, M.J.; Miller, M. Analysis of pesticides in dried hops by liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2008, 56, 6851–6856. [Google Scholar] [CrossRef]
- Manco, G.; Nucci, R.; Febbraio, F. Use of esterase activities for the detection of chemical neurotoxic agents. Protein Pept. Lett. 2009, 16, 1225–1234. [Google Scholar] [CrossRef]
- Mishra, R.K.; Dominguez, R.B.; Bhand, S.; Muñoz, R.; Marty, J.L. A novel automated flow-based biosensor for the determination of organophosphate pesticides in milk. Biosens. Bioelectron. 2012, 32, 56–61. [Google Scholar] [CrossRef]
- Pundir, C.S.; Chauhan, N. Acetylcholinesterase inhibition-based biosensors for pesticide determination: A review. Anal. Biochem. 2012, 429, 19–31. [Google Scholar] [CrossRef]
- Li, X.; Zheng, Z.; Liu, X.; Zhao, S.; Liu, S. Nanostructured photoelectrochemical biosensor for highly sensitive detection of organophosphorous pesticides. Biosens. Bioelectron. 2015, 64, 1–5. [Google Scholar] [CrossRef]
- Guler, M.; Turkoglu, V.; Kivrak, A. Electrochemical detection of malathion pesticide using acetylcholinesterase biosensor based on glassy carbon electrode modified with conducting polymer film. Environ. Sci. Pollut. Res. Int. 2016, 23, 12343–12351. [Google Scholar] [CrossRef]
- Febbraio, F.; D’Andrea, S.E.; Mandrich, L.; Merone, L.; Rossi, M.; Nucci, R.; Manco, G. Irreversible inhibition of the thermophilic esterase EST2 from Alicyclobacillus acidocaldarius. Extremophiles 2008, 12, 719–728. [Google Scholar] [CrossRef]
- Febbraio, F.; Merone, L.; Cetrangolo, G.P.; Rossi, M.; Nucci, R.; Manco, G. Thermostable esterase 2 from Alicyclobacillus acidocaldarius as biosensor for the detection of organophosphate pesticides. Anal. Chem. 2011, 83, 1530–1536. [Google Scholar] [CrossRef]
- Carullo, P.; Cetrangolo, G.P.; Mandrich, L.; Manco, G.; Febbraio, F. Fluorescence spectroscopy approaches for the development of a real-time organophosphate detection system using an enzymatic sensor. Sensors 2015, 15, 3932–3951. [Google Scholar] [CrossRef]
- Carullo, P.; Chino, M.; Cetrangolo, G.P.; Terreri, S.; Lombardi, A.; Manco, G.; Cimmino, A.; Febbraio, F. Direct detection of organophosphate compounds in water by a fluorescence-based biosensing device. Sens. Actuators B 2018, 255, 3257–3266. [Google Scholar] [CrossRef]
- Manco, G.; Adinolfi, E.; Pisani, F.M.; Ottolina, G.; Carrea, G.; Rossi, M. Overexpression and properties of a new thermophilic and thermostable esterase from Bacillus acidocaldarius with sequence similarity to hormone-sensitive lipase subfamily. Biochem. J. 1998, 332, 203–212. [Google Scholar] [CrossRef] [Green Version]
- De Simone, G.; Mandrich, L.; Menchise, V.; Giordano, V.; Febbraio, F.; Rossi, M.; Pedone, C.; Manco, G. A substrate-induced switch in the reaction mechanism of a thermophilic esterase—Kinetic evidences and structural basis. J. Biol. Chem. 2004, 279, 6815–6823. [Google Scholar] [CrossRef]
- Roberts, I.M. Hydrolysis of 4-methylumbelliferyl butyrate: A convenient and sensitive fluorescent assay for lipase activity. Lipids 1985, 20, 243–247. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Sotiropoulou, S.; Fournier, D.; Chaniotakis, N.A. Genetically engineered acetylcholinesterase-based biosensor for attomolar detection of dichlorvos. Biosens. Bioelectron. 2005, 20, 2347–2352. [Google Scholar] [CrossRef]
- Grange, J.M. A fluorigenic substrate for the rapid differentiation of Mycobacterium fortuitum from Mycobacterium chelonei on the basis of heat stable esterase activity. Tubercle 1977, 58, 147–150. [Google Scholar] [CrossRef]
- Mandrich, L.; Menchise, V.; Alterio, V.; De Simone, G.; Pedone, C.; Rossi, M.; Manco, G. Functional and structural features of the oxyanion hole in a thermophilic esterase from Alicyclobacillus acidocaldarius. Proteins 2008, 71, 1721–1731. [Google Scholar] [CrossRef]
- Dulaurent, S.; Moesch, C.; Marquet, P.; Gaulier, J.M.; Lachâtre, G. Screening of pesticides in blood with liquid chromatography-linear ion trap mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 2235–2249. [Google Scholar] [CrossRef]
- Berman, T.; Goldsmith, R.; Göen, T.; Spungen, J.; Novack, L.; Levine, H.; Amitai, Y.; Shohat, T.; Grotto, I. Urinary concentrations of organophosphate pesticide metabolites in adults in Israel: Demographic and dietary predictors. Environ. Int. 2013, 60, 183–189. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cetrangolo, G.P.; Gori, C.; Rusko, J.; Terreri, S.; Manco, G.; Cimmino, A.; Febbraio, F. Determination of Picomolar Concentrations of Paraoxon in Human Urine by Fluorescence-Based Enzymatic Assay. Sensors 2019, 19, 4852. https://doi.org/10.3390/s19224852
Cetrangolo GP, Gori C, Rusko J, Terreri S, Manco G, Cimmino A, Febbraio F. Determination of Picomolar Concentrations of Paraoxon in Human Urine by Fluorescence-Based Enzymatic Assay. Sensors. 2019; 19(22):4852. https://doi.org/10.3390/s19224852
Chicago/Turabian StyleCetrangolo, Giovanni Paolo, Carla Gori, Janis Rusko, Sara Terreri, Giuseppe Manco, Amelia Cimmino, and Ferdinando Febbraio. 2019. "Determination of Picomolar Concentrations of Paraoxon in Human Urine by Fluorescence-Based Enzymatic Assay" Sensors 19, no. 22: 4852. https://doi.org/10.3390/s19224852