
Analysis of Scat for Gut Microbiome Identification in Wolves from a Mediterranean and an Alpine Area

 , , , ,
, , , , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
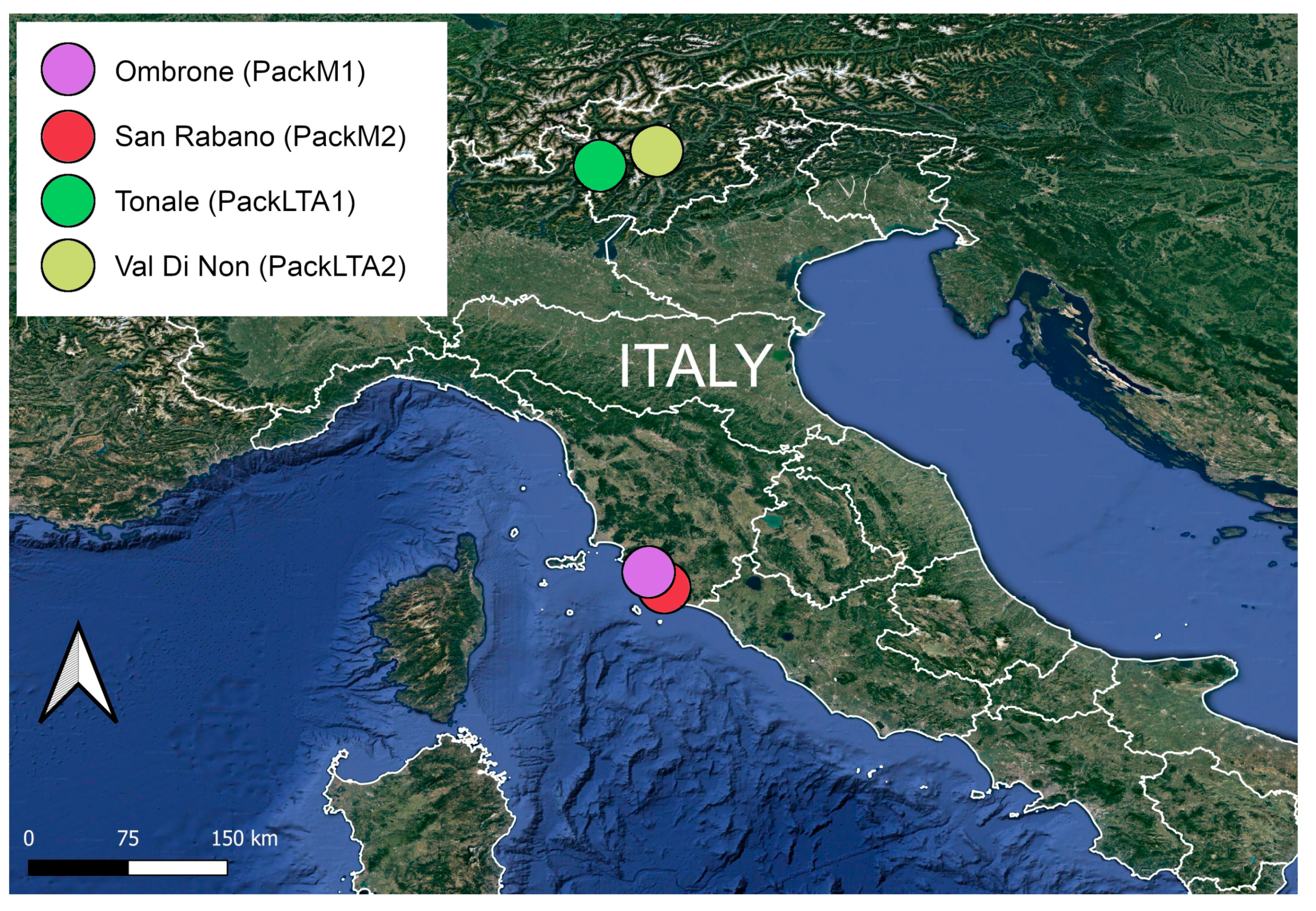
2.1. Study Areas
2.2. Sample Collection and Assessment of Scat Content
2.3. Molecular Analyses
2.4. Alpha and Beta Diversity of Gut Microbiome
2.5. Testing for Differences among Packs
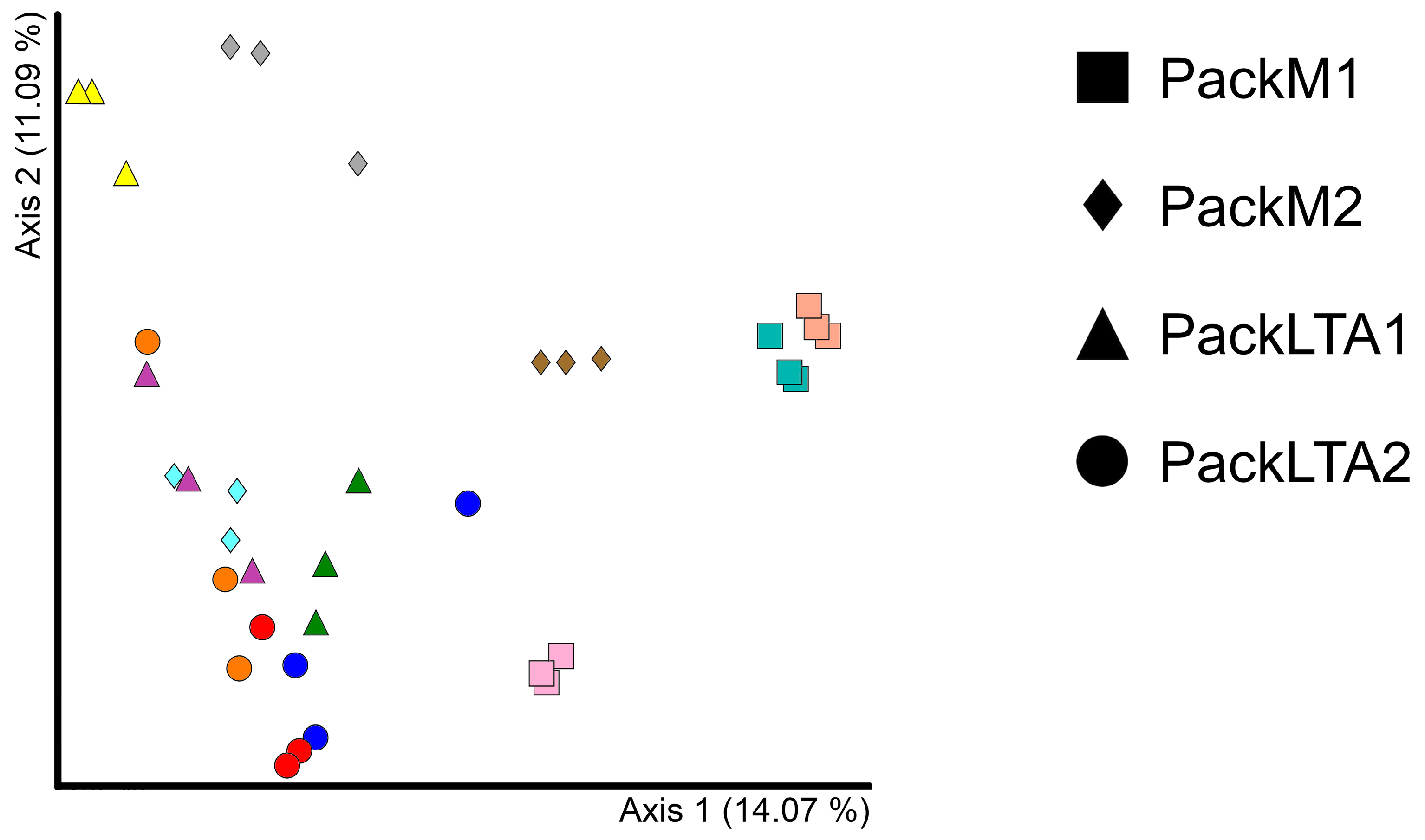
3. Results
3.1. Scat Content
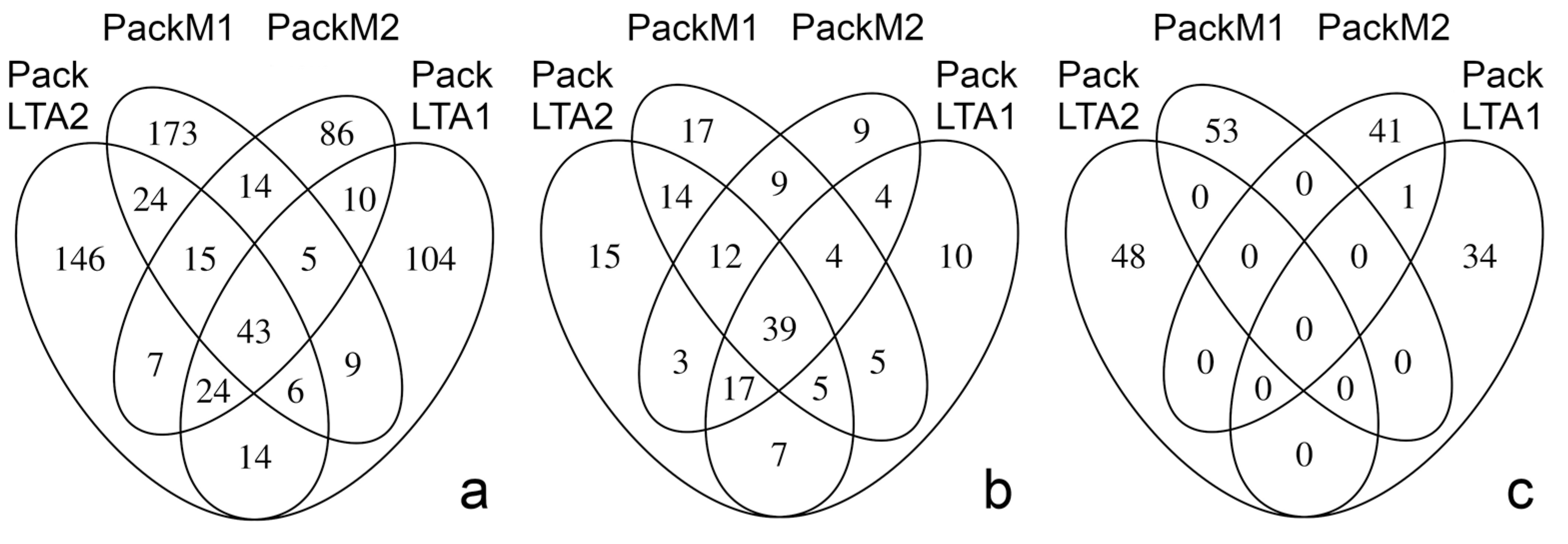
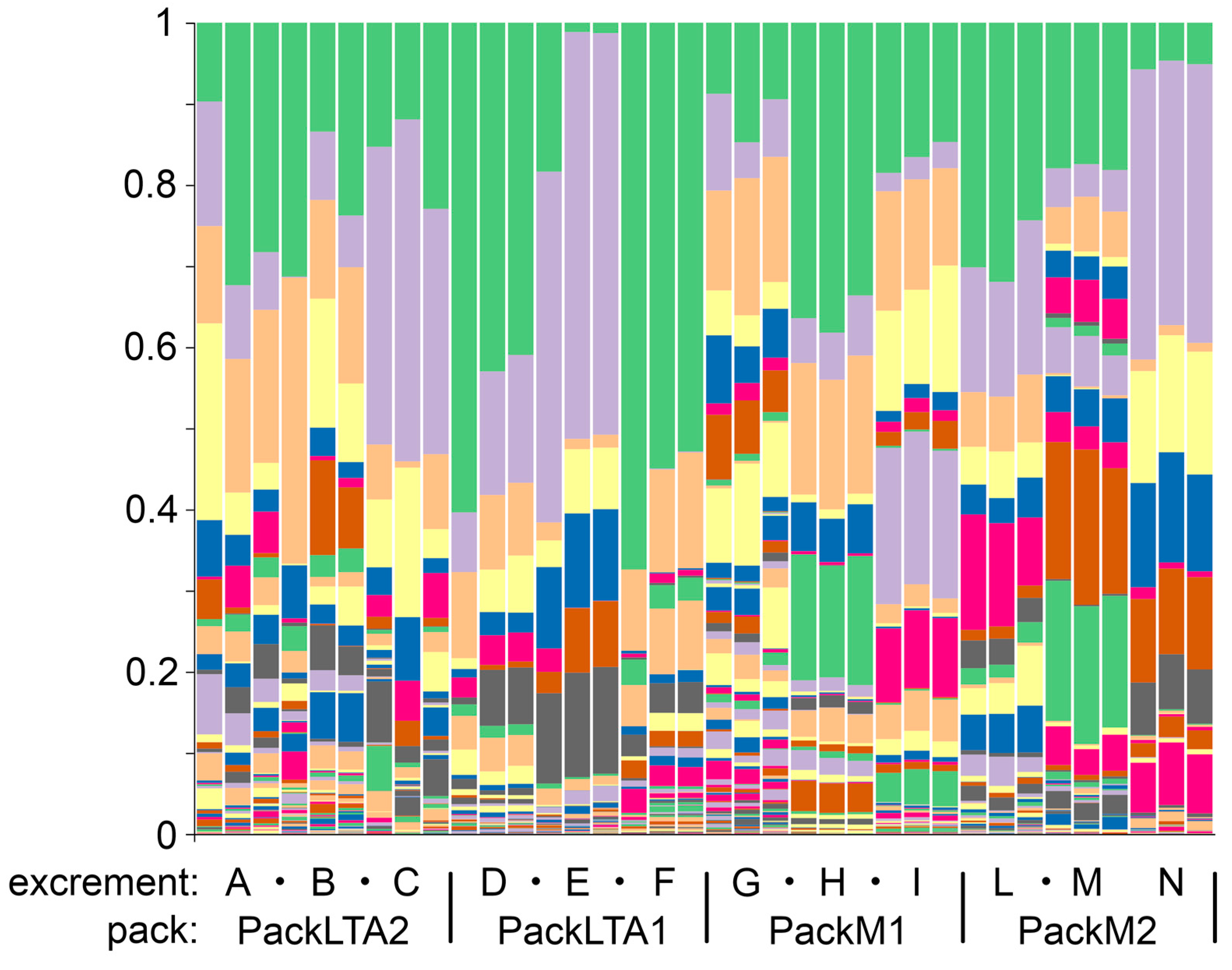
3.2. Microbial Diversity in Excrements
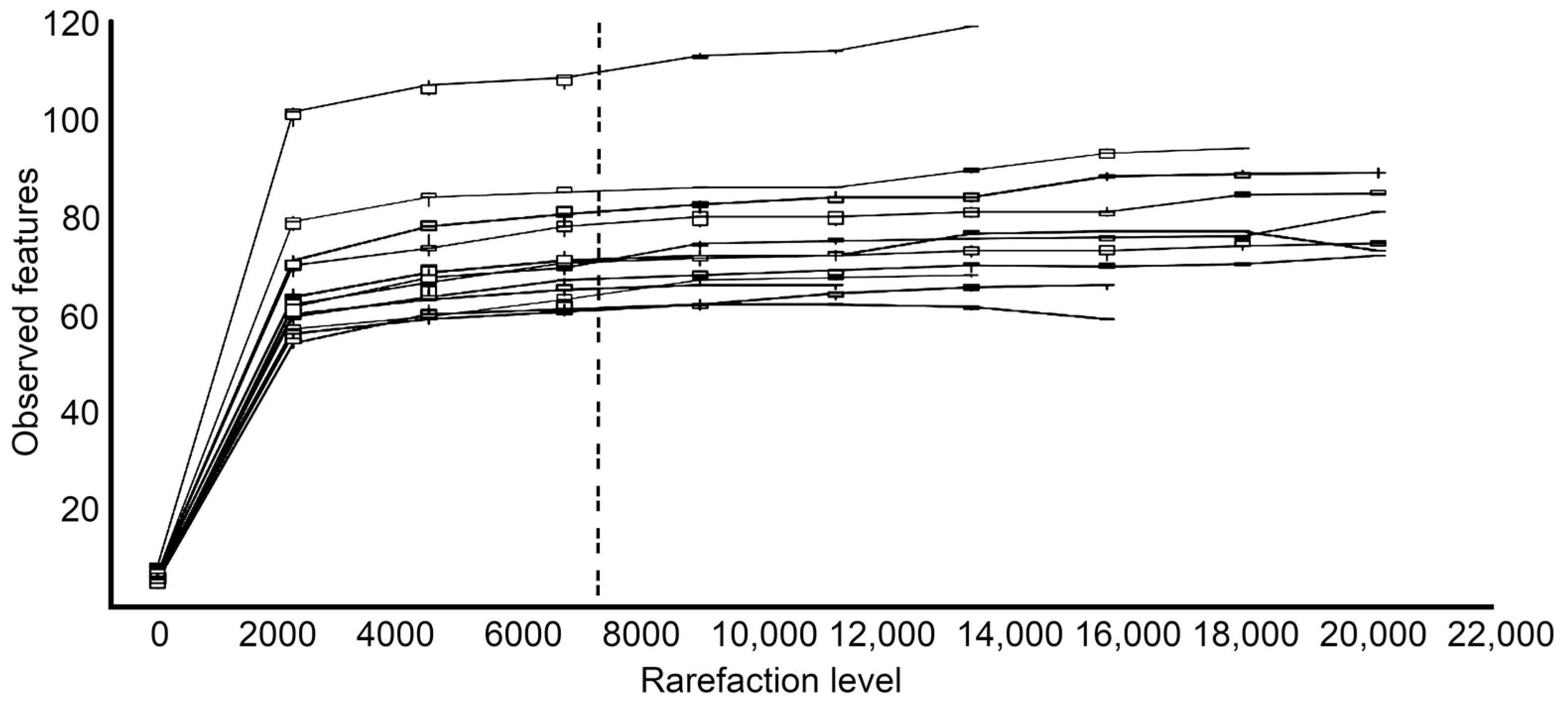
3.3. Alpha and Beta Diversity
3.4. Testing for Differences at Different Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular analysis of commensal host–microbial relationships in the intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.H.; Ochman, H. Rates of gut microbiome divergence in mammals. Mol. Ecol. 2018, 27, 1884–1897. [Google Scholar] [CrossRef]
- Menke, S.; Meier, M.; Sommer, S. Shifts in the gut microbiome observed in wildlife faecal samples exposed to natural weather conditions: Lessons from time-series analyses using next-generation sequencing for application in field studies. Methods Ecol. Evol. 2015, 6, 1080–1087. [Google Scholar] [CrossRef]
- Kartzinel, T.R.; Hsing, J.C.; Musili, P.M.; Brown, B.R.; Pringle, R.M. Covariation of diet and gut microbiome in African megafauna. Proc. Natl. Acad. Sci. USA 2019, 116, 23588–23593. [Google Scholar] [CrossRef] [Green Version]
- Pannoni, S.B.; Proffitt, K.M.; Holben, W.E. Non-invasive monitoring of multiple wildlife health factors by fecal microbiome analysis. Ecol. Evol. 2022, 12, e8564. [Google Scholar] [CrossRef]
- Sugden, S.; St Clair, C.C.; Stein, L.Y. Individual and site-specific variation in a biogeographical profile of the coyote gastrointestinal microbiota. Microb. Ecol. 2020, 81, 240–252. [Google Scholar] [CrossRef]
- Bird, S.; Prewer, E.; Kutz, S.; Leclerc, L.-M.; Vilaça, S.T.; Kyle, C.J. Geography, seasonality, and host-associated population structure influence the fecal microbiome of a genetically depauparate Arctic mammal. Ecol. Evol. 2019, 9, 13202–13217. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Sun, Y.; Zhu, F.; Liu, Z.; Pan, R.; Teng, L.; Guo, S. Seasonal Variation and Sexual Dimorphism of the Microbiota in Wild Blue Sheep (Pseudois nayaur). Front. Microbiol. 2020, 11, 1260. [Google Scholar] [CrossRef]
- Boitani, L.; Phillips, M.; Jhala, Y. Canis lupus. The IUCN Red List of Threatened Species; IUCN: Gland, Switzerland, 2018; p. e.T3746A119623865. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Chen, J.; Shang, S.; Yan, J.; Chen, Y.; Tang, X.; Zhang, H. Analysis and comparison of the wolf microbiome under different environmental factors using three different data of Next Generation Sequencing. Sci. Rep. 2017, 7, 11332. [Google Scholar] [CrossRef] [Green Version]
- DeCandia, A.L.; Cassidy, K.A.; Stahler, D.R.; Stahler, E.A.; VonHoldt, B.M. Social environment and genetics underlie body site specific microbiomes of Yellowstone National Park gray wolves (Canis lupus). Ecol. Evol. 2021, 11, 9472–9488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, L. Phylogenetic analysis of 16S rRNA gene sequences reveals distal gut bacterial diversity in wild wolves (Canis lupus). Mol. Biol. Rep. 2010, 37, 4013–4022. [Google Scholar] [CrossRef] [PubMed]
- Mech, L.D.; Boitani, L. Wolf social ecology. In Wolves: Behavior, Ecology, and Conservation; Mech, L.D., Boitani, L., Eds.; University of Chicago Press: Chicago, IL, USA, 2003. [Google Scholar]
- Ferretti, F.; Lovari, S.; Mancino, V.; Burrini, L.; Rossa, M. Food habits of wolves and selection of wild ungulates in a prey-rich Mediterranean coastal area. Mamm. Biol. 2019, 99, 119–127. [Google Scholar] [CrossRef]
- Kruuk, H.; Parish, T. Feeding specialization of the European badger Meles meles in Scotland. J. Anim. Ecol. 1981, 50, 773–788. [Google Scholar] [CrossRef]
- Ferretti, F.; Pacini, G.; Belardi, I.; Ten Cate, B.; Sensi, M.; Oliveira, R.; Rossa, M.; Burrini, L.; Lovari, S. Recolonizing wolves and opportunistic foxes: Interference or facilitation? Biol. J. Linn. Soc. 2021, 132, 196–210. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 15, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Anderson, M.J.; Gorley, R.N.; Clarke, K.R. PERMANOVA + for PRIMER: Guide to Software and Statistical Methods; PRIMER-E: Auckland, New Zealand, 2008. [Google Scholar]
- Bragg, M.; Freeman, E.W.; Lim, H.C.; Songsasen, N.; Muletz-Wolz, C.R. Gut microbiomes differ among dietary types and stool consistency in the captive red wolf (Canis rufus). Front. Microbiol. 2020, 11, 590212. [Google Scholar] [CrossRef]
- Colborn, A.S.; Kuntze, C.C.; Gadsden, G.I.; Harris, N.C. Spatial variation in diet–microbe associations across populations of a generalist North American carnivore. J. Anim. Ecol. 2020, 89, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- DeCandia, A.L.; Brenner, L.J.; King, J.L.; vonHoldt, B.M. Ear mite infection is associated with altered microbial communities in genetically depauperate Santa Catalina Island foxes (Urocyon littoralis catalinae). Mol. Ecol. 2020, 29, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- DeCandia, A.L.; Leverett, K.N.; Vonholdt, B.M. Of microbes and mange: Consistent changes in the skin microbiome of three canid species infected with Sarcoptes scabiei mites. Parasit. Vectors 2019, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Hoffmann, A.; Patterson, A.P.; Diesel, A.; Lawhon, S.D.; Ly, H.J.; Elkins Stephenson, C.; Masnell, J.; Steiner, J.M.; Dowd, S.E.; Olivry, T.; et al. The Skin Microbiome in Healthy and Allergic Dogs. PLoS ONE 2014, 9, e83197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, T.M.; Rogers, T.L.; Brown, M.V. The gut bacterial community of mammals from marine and terrestrial habitats. PLoS ONE 2013, 8, e83655. [Google Scholar] [CrossRef]
- Roggenbuck, M.; Schnell, I.B.; Blom, N.; Bælum, J.; Bertelsen, M.F.; Sicheritz-Ponten, T.; Hansen, L.H. The microbiome of New World vultures. Nat. Commun. 2014, 5, 5498. [Google Scholar] [CrossRef] [Green Version]
- Bermingham, E.N.; Maclean, P.; Thomas, D.G.; Cave, N.J.; Young, W. Key bacterial families (Clostridiaceae, Erysipelotrichaceae and Bacteroidaceae) are related to the digestion of protein and energy in dogs. PeerJ 2017, 5, e3019. [Google Scholar] [CrossRef] [Green Version]
- Pilla, R.; Suchodolski, J.S. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front. Vet. Sci. 2020, 6, 498. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Hyde, E.R.; Suchodolski, J.S.; Knight, R. Dog and human inflammatory bowel disease rely on overlapping yet distinct dysbiosis networks. Nat. Microbiol. 2016, 1, 16177. [Google Scholar] [CrossRef]
- Ross, A.A.; Müller, K.M.; Scott Weese, J.; Neufeld, J.D. Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc. Natl. Acad. Sci. USA 2018, 115, E5786–E5795. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2015, 9, 832–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattorini, N.; Ferretti, F. Estimating wild boar density and rooting activity in a Mediterranean protected area. Mamm. Biol. 2020, 100, 241–251. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Jaccard | Bray-Curtis | Unifrac | Weighted Unifrac | |
|---|---|---|---|---|
| Pack | 0.275 | 0.266 | 0.229 | 0.273 |
| Excrement | 0.464 | 0.624 | 0.535 | 0.608 |
| Residuals | 0.261 | 0.110 | 0.236 | 0.119 |
| Jaccard | Bray-Curtis | Unifrac | Weighted Unifrac | |
|---|---|---|---|---|
| PRIMER/PERMANOVA+ | ||||
| test statistic (packs) | 1.5773 | 1.1358 | 1.1422 | 1.1975 |
| p (packs) | 0.0346 * | 0.3615 | 0.3327 | 0.3247 |
| post hoc pairwise tests p (packs) | LTA2–LTA1: 0.144 LTA2–M1: 0.113 LTA2–M2: 0.227 LTA1–M1: 0.103 LTA1–M2: 0.266 M1–M2: 0.217 | N/A ** | N/A ** | N/A ** |
| test statistic (excrements) | 5.3344 | 17.039 | 6.8207 | 15.281 |
| p (excrements) | 0.0001 * | 0.0001 * | 0.0001 * | 0.0001 * |
| post hoc pairwise tests p (excrements) | all significant (p < 0.05) except two pairs in PackLTA2 | all significant (p < 0.05) except one pair in PackLTA2 | all significant (p < 0.05) except one pair in PackLTA2, one pair in PackLTA1 and all pairs in PackM2 | all significant (p < 0.05) except one pair in PackLTA2 |
| qiime diversity beta-group-significance (combining technical replicates) | ||||
| test statistic | 1.366 | 1.200 | 1.406 | 1.239 |
| p (packs) | 0.0011 * | 0.2481 | 0.1406 | 0.2701 |
| post hoc pairwise tests p (packs) | LTA2–LTA1: 0.105 LTA2–M1: 0.101 LTA2–M2: 0.097 LTA1–M1: 0.099 LTA1–M2: 0.266 M1–M2: 0.217 | N/A ** | N/A ** | N/A ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardi, F.; Lazzeri, L.; Iannotti, N.; Donini, V.; Cucini, C.; Belardi, I.; Frati, F.; Carapelli, A.; Ferretti, F. Analysis of Scat for Gut Microbiome Identification in Wolves from a Mediterranean and an Alpine Area. Diversity 2023, 15, 37. https://doi.org/10.3390/d15010037
Nardi F, Lazzeri L, Iannotti N, Donini V, Cucini C, Belardi I, Frati F, Carapelli A, Ferretti F. Analysis of Scat for Gut Microbiome Identification in Wolves from a Mediterranean and an Alpine Area. Diversity. 2023; 15(1):37. https://doi.org/10.3390/d15010037
Chicago/Turabian StyleNardi, Francesco, Lorenzo Lazzeri, Nicola Iannotti, Valerio Donini, Claudio Cucini, Irene Belardi, Francesco Frati, Antonio Carapelli, and Francesco Ferretti. 2023. "Analysis of Scat for Gut Microbiome Identification in Wolves from a Mediterranean and an Alpine Area" Diversity 15, no. 1: 37. https://doi.org/10.3390/d15010037