
Bacterial Community in the Gut of Neanthes japonica and Its Association with Surrounding Environment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.3. Sequencing Process and Statistical Analysis
3. Results
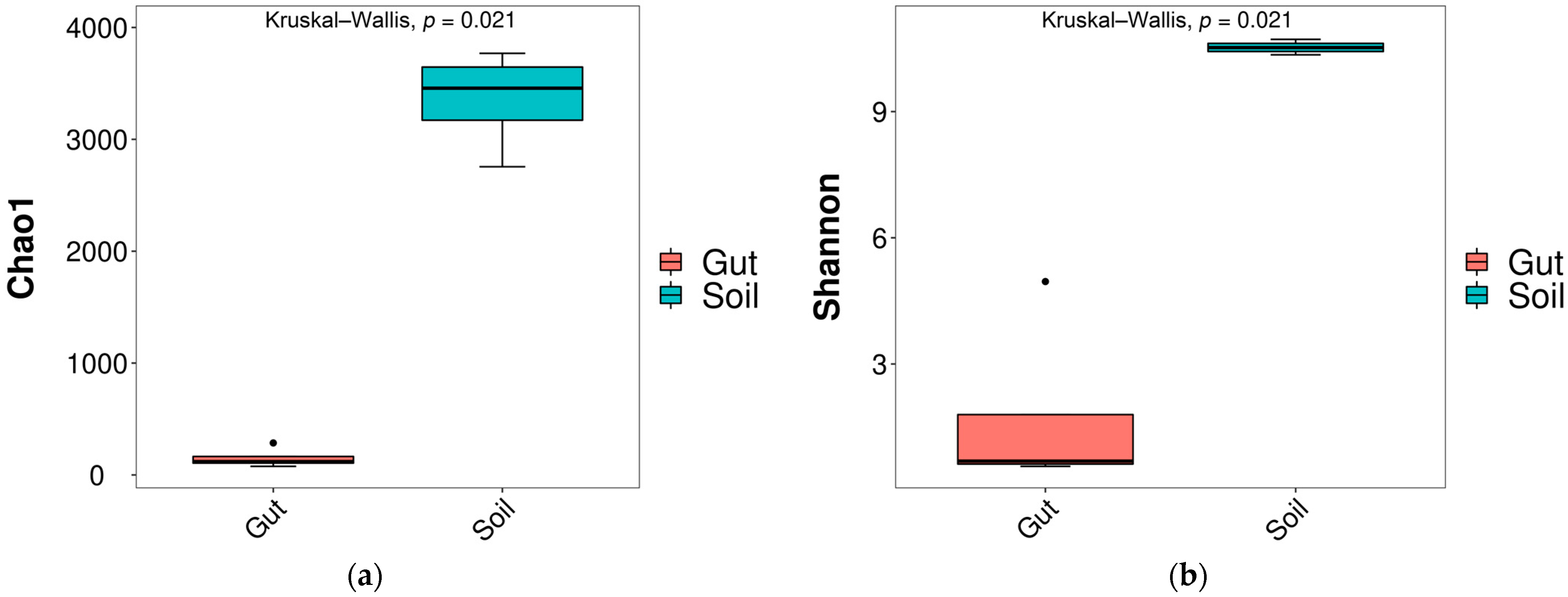
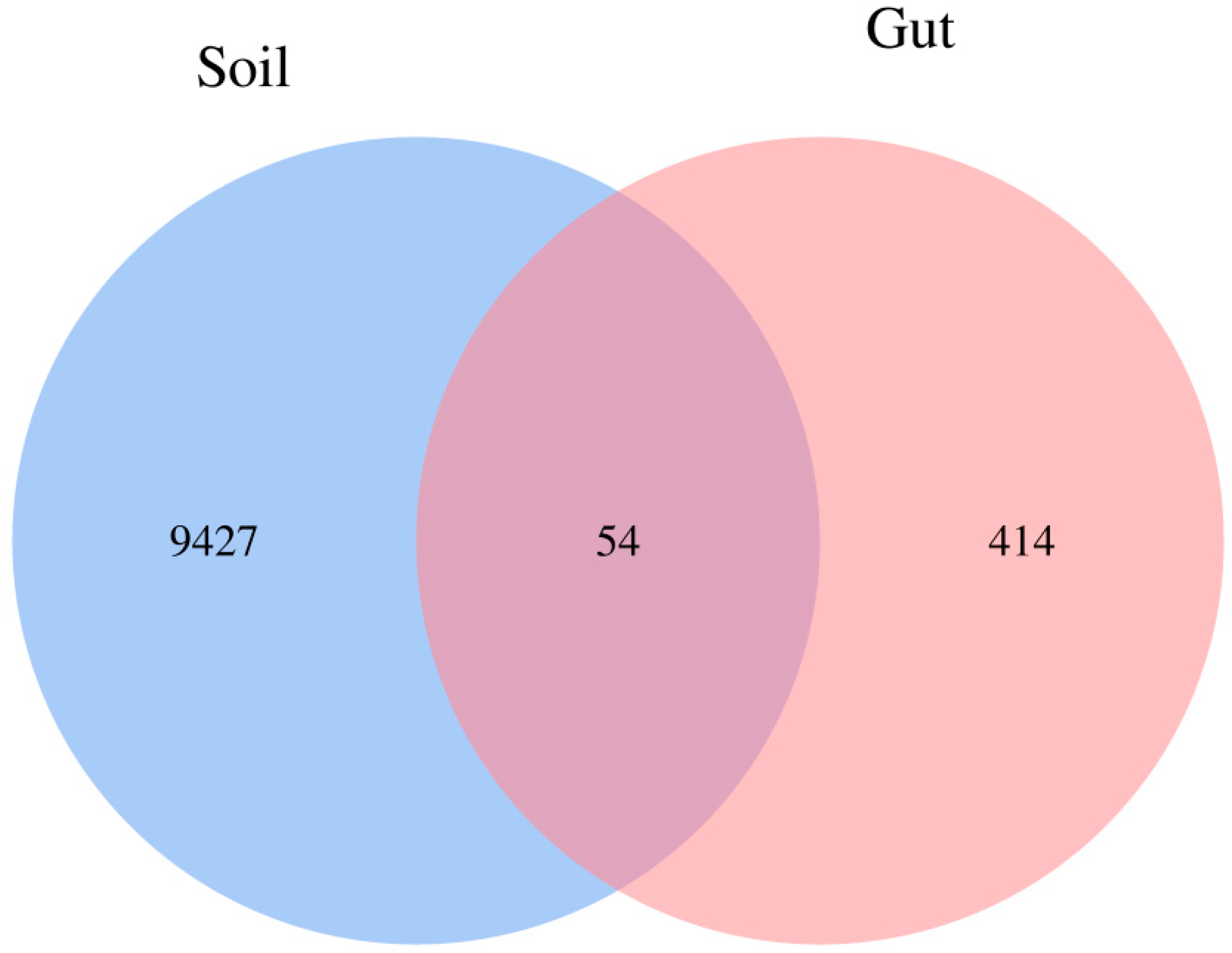
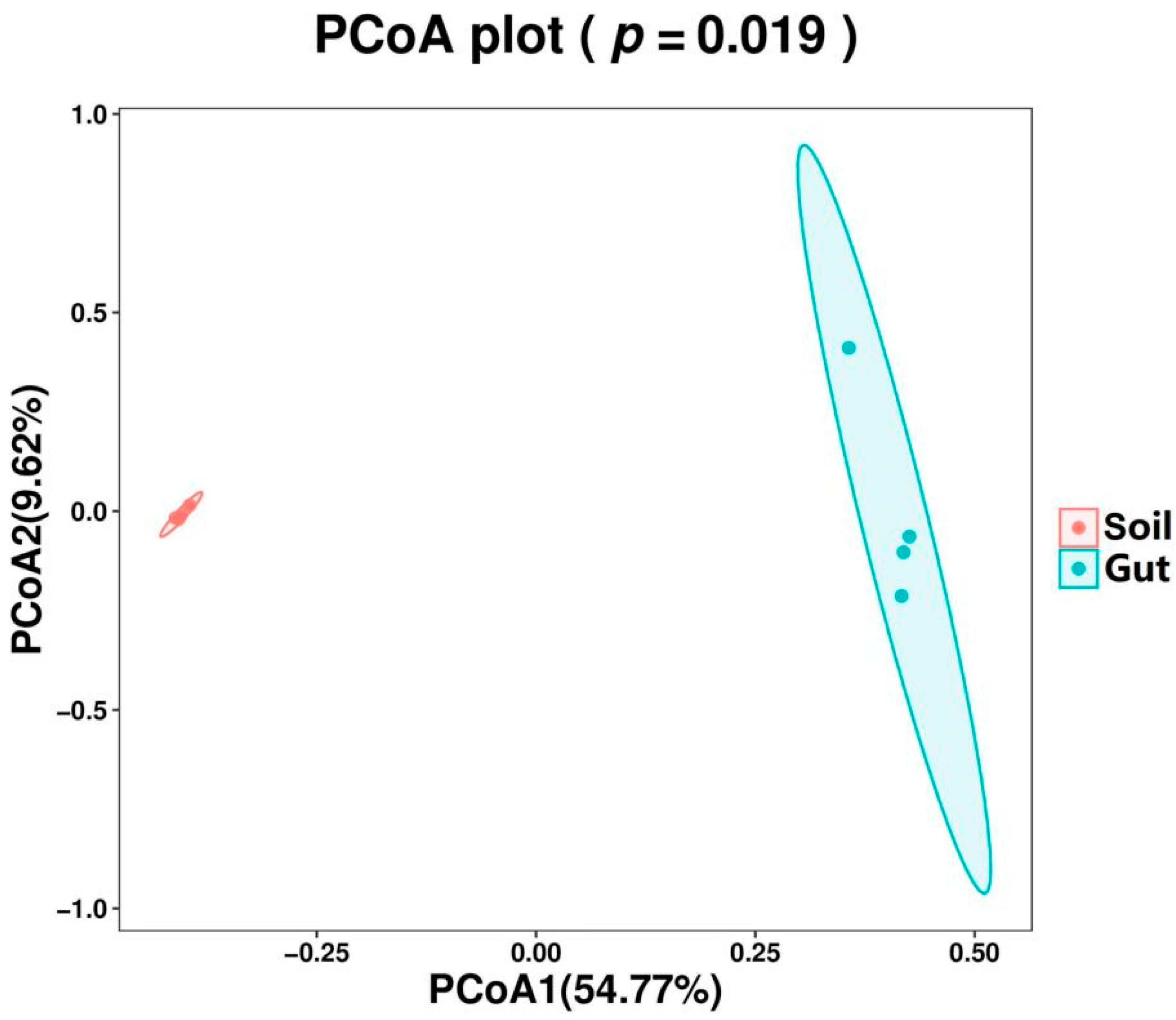
3.1. Bacterial Diversity
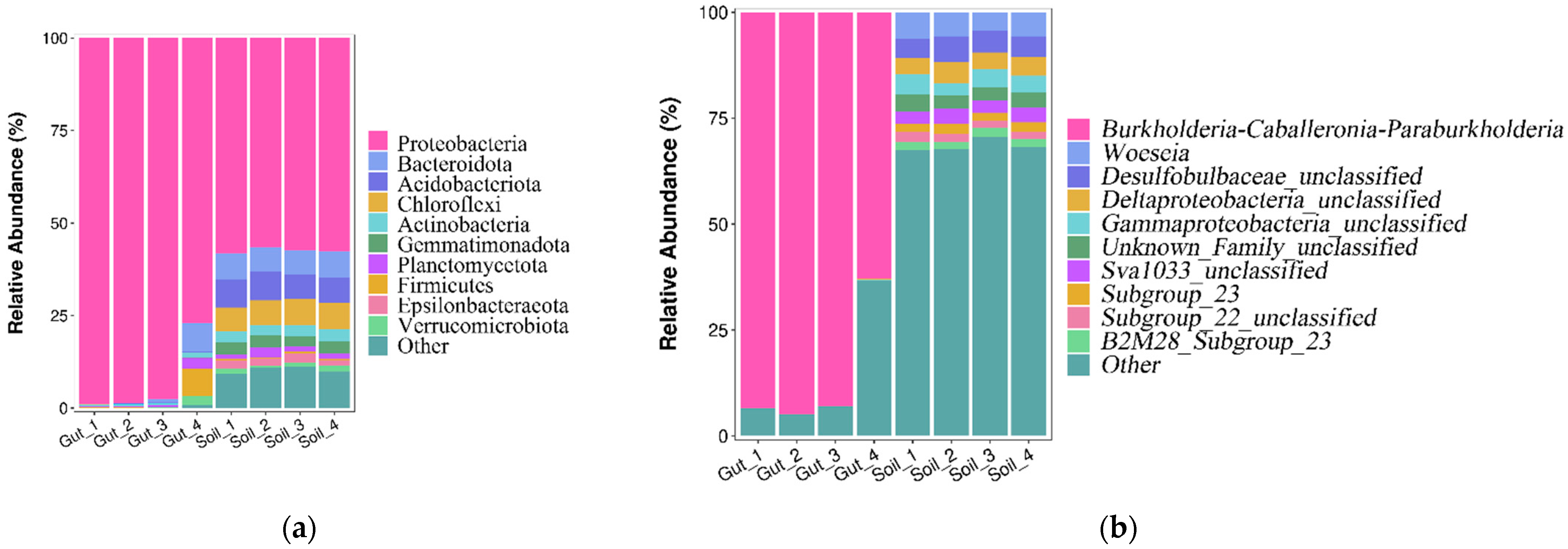
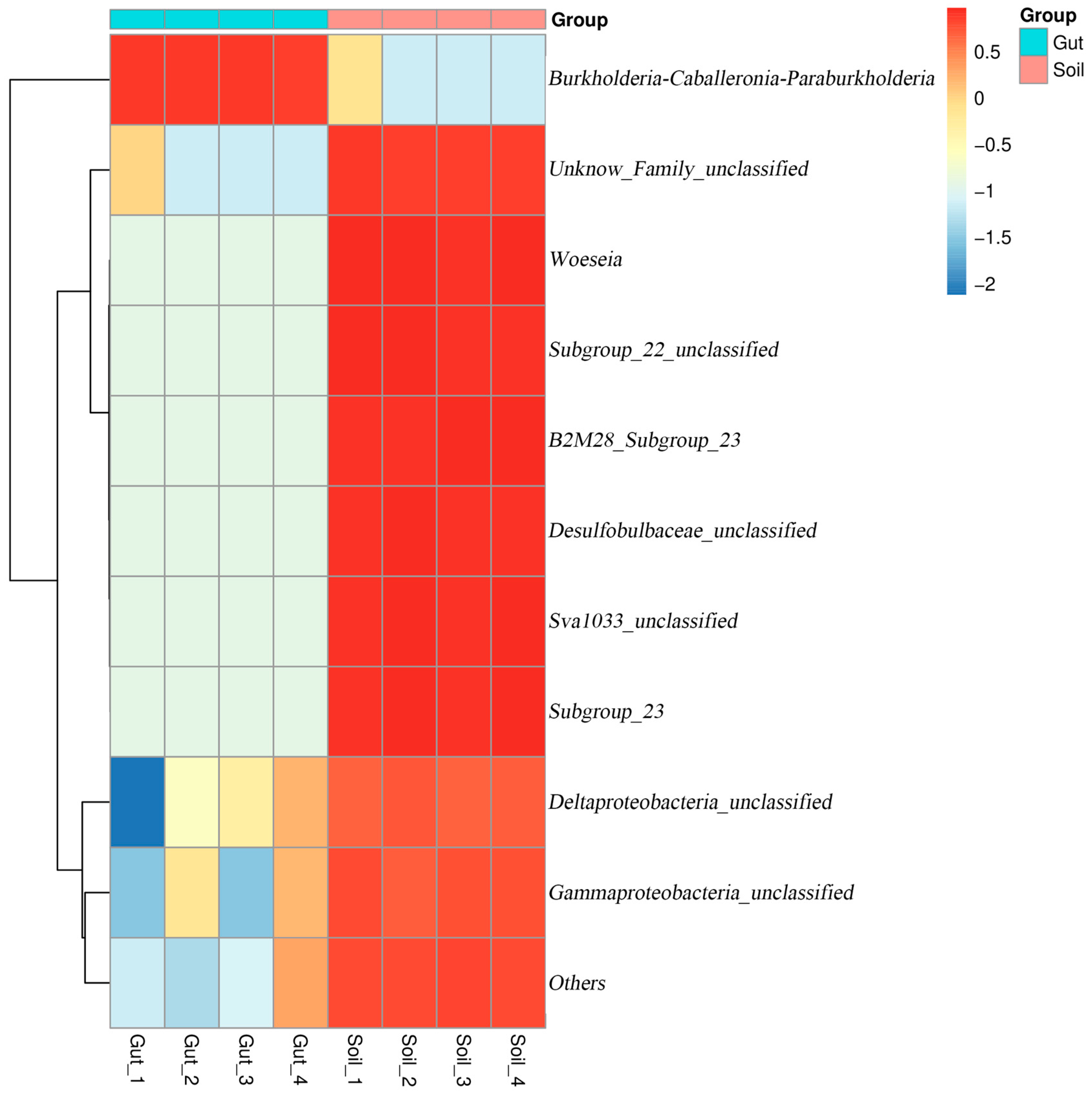
3.2. Bacterial Community Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, W.-J.; Hase, K. Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 2014, 10, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Ajuwon, K.M.; Fang, R. Mechanistic insight into the gut microbiome and its interaction with host immunity and inflammation. Anim. Nutr. 2020, 6, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Liu, B.; Wu, H.; Feng, J.; Jiang, T. Seasonal dietary shifts alter the gut microbiota of avivorous bats: Implication for adaptation to energy harvest and nutritional utilization. mSphere 2021, 6, e0046721. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Lu, T.; Yu, Y.; Penuelas, J.; Zhu, Y.-G.; Qian, H. Gammaproteobacteria, a core taxon in the guts of soil fauna, are potential responders to environmental concentrations of soil pollutants. Microbiome 2021, 9, 196. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Zhou, Z.; He, S.; Cao, Y.; Shi, P.; Yao, B.; Ringø, E. Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aquac. Res. 2010, 42, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Gong, L.; Khan, T.A.; Pan, L.; Yan, L.; Li, D.; Cao, L.; Li, Y.; Ding, X.; Yi, G.; et al. Comparative analysis and gut bacterial community assemblages of grass carp and crucian carp in new lineages from the Dongting Lake area. MicrobiologyOpen 2020, 9, e996. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, W.; Jiang, Y.; Dai, W.; Li, P.; Yao, D.; Wang, J.; Shi, Y.; Cui, Z.; Cao, H.; et al. Variations in bacterial taxonomic profiles and potential functions in response to the gut transit of earthworms (Eisenia fetida) feeding on cow manure. Sci. Total Environ. 2021, 787, 147392. [Google Scholar] [CrossRef]
- Liu, Y.; Xian, W. The effect of temperature on growth and energy budget of the polychaete, Neanthes japonica Izuka. J. Ocean Univ. China 2009, 8, 177–183. [Google Scholar] [CrossRef]
- Ye, J. Biological Characteristics of Neanthes japonica and their application in shrimp aquaculture. J. Anhui Agric. Sci. 2010, 48, 7883–7938. [Google Scholar]
- Wang, S.; Deng, Z.; Li, Q.; Ge, X.; Bo, Q.; Liu, J.; Cui, J.; Jiang, X.; Liu, J.; Zhang, L.; et al. A novel alkaline serine protease with fibrinolytic activity from the polychaete, Neanthes japonica. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2011, 159, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, S.; Li, Q.; Ji, X.; Zhang, L.; Hong, M. Purification and characterization of a novel fibrinolytic enzyme from the polychaete, Neanthes japonica (Iznka). Bioresour. Technol. 2010, 101, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.B.; Stedmon, C.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindström, E.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler dia-grams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Wagner, H.H. Vegan Community Ecology Package Version 2.5-7 November; R. Foundation: Vienna, Austria, 2020. [Google Scholar]
- Rungrassamee, W.; Klanchui, A.; Chaiyapechara, S.; Maibunkaew, S.; Tangphatsornruang, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Bacterial Population in Intestines of the Black Tiger Shrimp (Penaeus monodon) under Different Growth Stages. PLoS ONE 2013, 8, e60802. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Guan, W.; Wei, G.; Liu, B.; Xu, J.; Zhao, L.; Zhang, Y. Phylogenetic analysis of intestinal bacteria in the Chinese mitten crab (Eriocheir sinensis). J. Appl. Microbiol. 2007, 103, 675–682. [Google Scholar] [CrossRef]
- Sun, Z.; Xuan, Y.; Zhang, H.; Jiang, M.; Pan, Y.; Zhang, Y.; Gong, Y.; Lu, X.; Yu, D.; Xue, R.; et al. Bacterial diversity in the Penaeus vannamei Boone intestine and aquaculture environment. J. Fish. Sci. China 2016, 23, 594–605. [Google Scholar]
- Chaiyapechara, S.; Rungrassamee, W.; Suriyachay, I.; Kuncharin, Y.; Klanchui, A.; Karoonuthaisiri, N.; Jiravanichpaisal, P. Bacterial Community Associated with the Intestinal Tract of P. monodon in Commercial Farms. Microb. Ecol. 2012, 63, 938–953. [Google Scholar] [CrossRef]
- Liu, D.; Lian, B.; Wu, C.; Guo, P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis 2017, 74, 21–29. [Google Scholar] [CrossRef]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xu, D.; Zhu, J.; Wang, S.; Liu, M.; Sun, M.; Wang, G.; Song, L.; Liu, X.; Xie, T. Habitat environmental factors influence intestinal microbial diversity of the short-faced moles (Scaptochirus moschata). AMB Express 2021, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Liu, M.; Wang, B.; Jiang, K.; Ma, S.; Li, Q. The intestinal microbial diversity in Chinese shrimp (Fenneropenaeus chinensis) as determined by PCR–DGGE and clone library analyses. Aquaculture 2011, 317, 32–36. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Wu, P.; Chen, M.; He, L.; Xu, Q.; Wang, A. Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers (Holothuria atra and H. leucospilota). J. Oceanol. Limnol. 2021, 40, 360–372. [Google Scholar] [CrossRef]
- Zhang, B.; Lian, B.; Wang, B.; Zhou, Y.; He, J. PCR-DGGE analysis of immobilized microbial diversity in digestive tract of sand worm Perinereis aibuhitensis. J. Dalian Ocean. Univ. 2013, 28, 413–417. [Google Scholar]
- Hu, Y.; Xie, H.; Gao, M.; Huang, P.; Zhou, H.; Ma, Y.; Zhou, M.; Liang, J.; Yang, J.; Lv, Z. Dynamic of Composition and Diversity of Gut Microbiota in Triatoma rubrofasciata in Different Developmental Stages and Environmental Conditions. Front. Cell. Infect. Microbiol. 2020, 10, 587708. [Google Scholar] [CrossRef]
- Zla, B.; Sp, A.; Zhe, Z.A.; Xw, A.; Jla, B.; Yha, B.; Wza, B.; Qla, B.; Pba, B.; Msa, B. Novel pathway of acephate degra-dation by the microbial consortium ZQ01 and its potential for environmental bioremediation. J. Hazard. Mater. 2021, 426, 127841. [Google Scholar]
- Ge, L. Advances in the study of bacteria in intestines of fishes. Reserv. Fish. 2006, 26, 17–20. [Google Scholar]
- Jin, R.; Jiang, M.; Sun, S.; Dai, X.; Wu, H.; Zhou, J.; Yu, Z.; Zhang, F. Microbial community in Litopenaeus vannamei intestine and its aquaculture environment. J. Fish. China 2020, 44, 2037–2054. [Google Scholar]
- Zhang, Z.; Li, B.; Wang, Y.; Liao, M.; Wang, L.; Rong, X. The microflora structure in digestive tract of half-smooth tongue sole (Cynoglossus semilaevis Gunther) cultured in outdoor pond basing on high-through sequencing technique. Acta Hydrobiol. Sin. 2015, 39, 38–45. [Google Scholar]
- Wang, X.; Zhao, Y.; Song, Z.; Zhong, S.; Huang, G.; Tong, T.; Nie, Z.; Su, Q.; Yang, J. Application of high-throughput sequencing techniques for analyzing bacterial communities in pond-raised mud crab (Scylla paramamosain) intestine and its aquaculture environment. J. Fish. Sci. China 2017, 24, 1245. [Google Scholar] [CrossRef]
- Fang, A.Q.; He, Z.L.; Wang, C.; Yang, C.; Yan, Q.Y. Progress in studying microbially-driven sulfur cycling in man-grove sediments. Acta Microbiol. Sin. 2020, 60, 13–25. [Google Scholar]
- Li, Z.; Liu, L. Advance of study in Delftia. China Trop. Med. 2008, 18, 2254–2255. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, S.; Li, L.; Liu, X.; Wang, J.; Tang, X. Bacterial Community in the Gut of Neanthes japonica and Its Association with Surrounding Environment. Diversity 2022, 14, 514. https://doi.org/10.3390/d14070514
Shang S, Li L, Liu X, Wang J, Tang X. Bacterial Community in the Gut of Neanthes japonica and Its Association with Surrounding Environment. Diversity. 2022; 14(7):514. https://doi.org/10.3390/d14070514
Chicago/Turabian StyleShang, Shuai, Liangyu Li, Xiaoxue Liu, Jun Wang, and Xuexi Tang. 2022. "Bacterial Community in the Gut of Neanthes japonica and Its Association with Surrounding Environment" Diversity 14, no. 7: 514. https://doi.org/10.3390/d14070514