

The Quality of Sequence Data Affects Biodiversity and Conservation Perspectives in the Neotropical Damselfly Megaloprepus caerulatus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Mining
2.2. Sequence Editing and Alignments
- (i)
- Alignments 1 include the original Study-A sequences (for CO1 and 16S rDNA separately),
- (ii)
- Alignments 2 consist of the edited Alignments 1 (again, separately for CO1 and 16S rDNA), and
- (iii)
- Alignments 3 include the combined sequence data from both studies (Alignments 2 from Study-A plus Study-B alignments for either CO1 or 16S rDNA).
2.3. Population Structure, Genealogical Network, and Phylogenetic Analyses
3. Results
3.1. Alignments
3.1.1. 16S
3.1.2. CO1
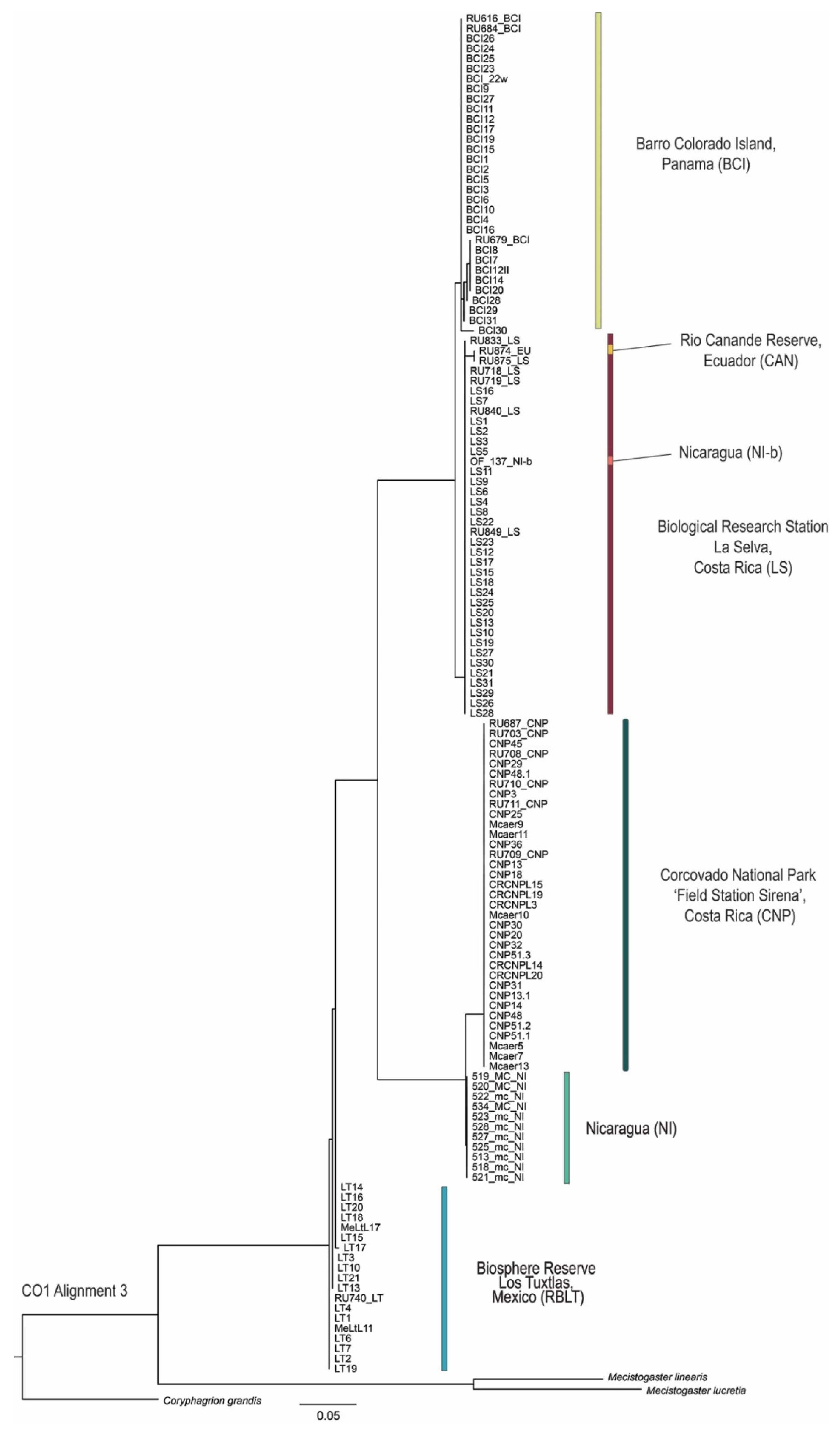
3.2. Population Structure, Genealogical Network, and Phylogenetic Analyses
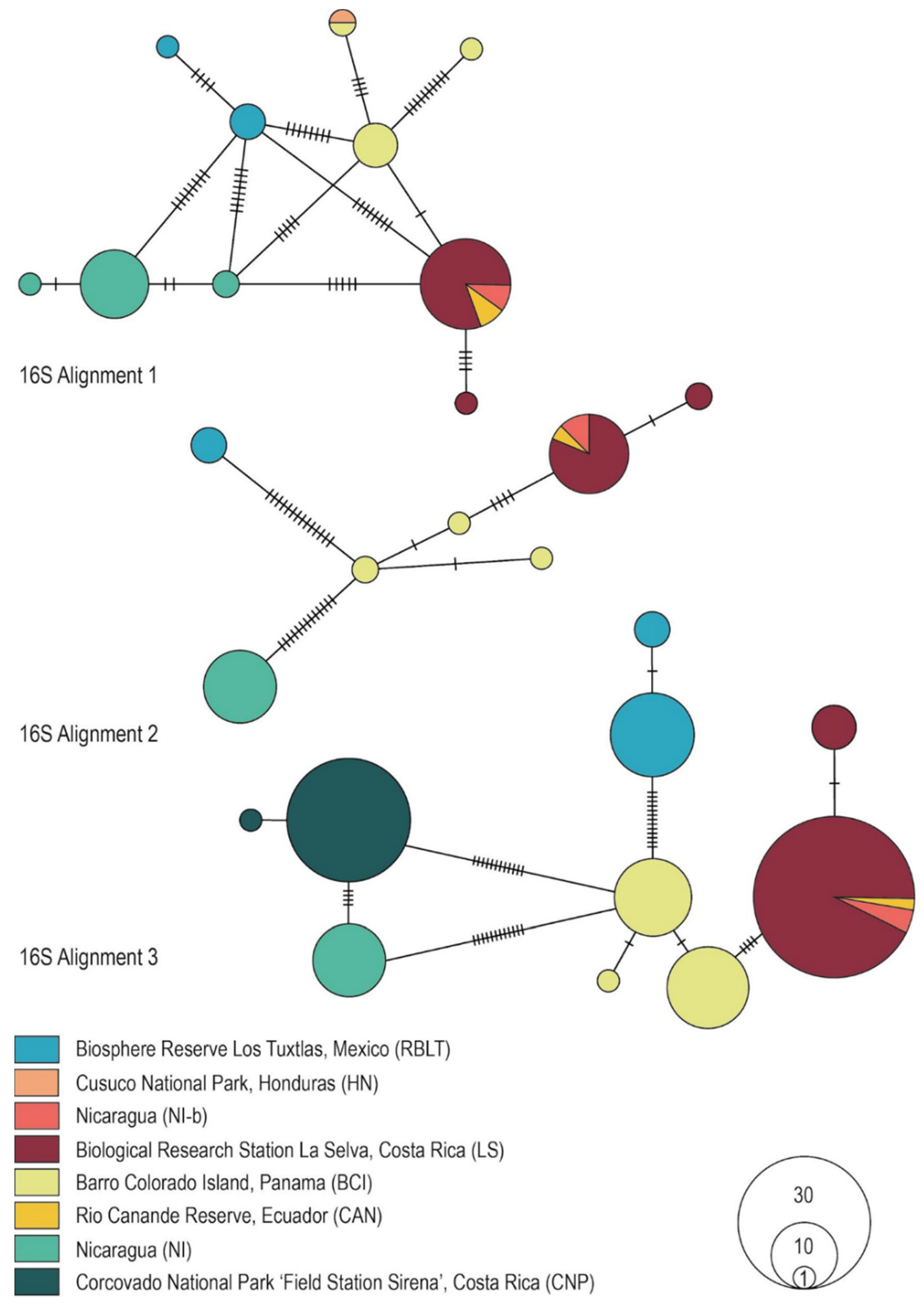
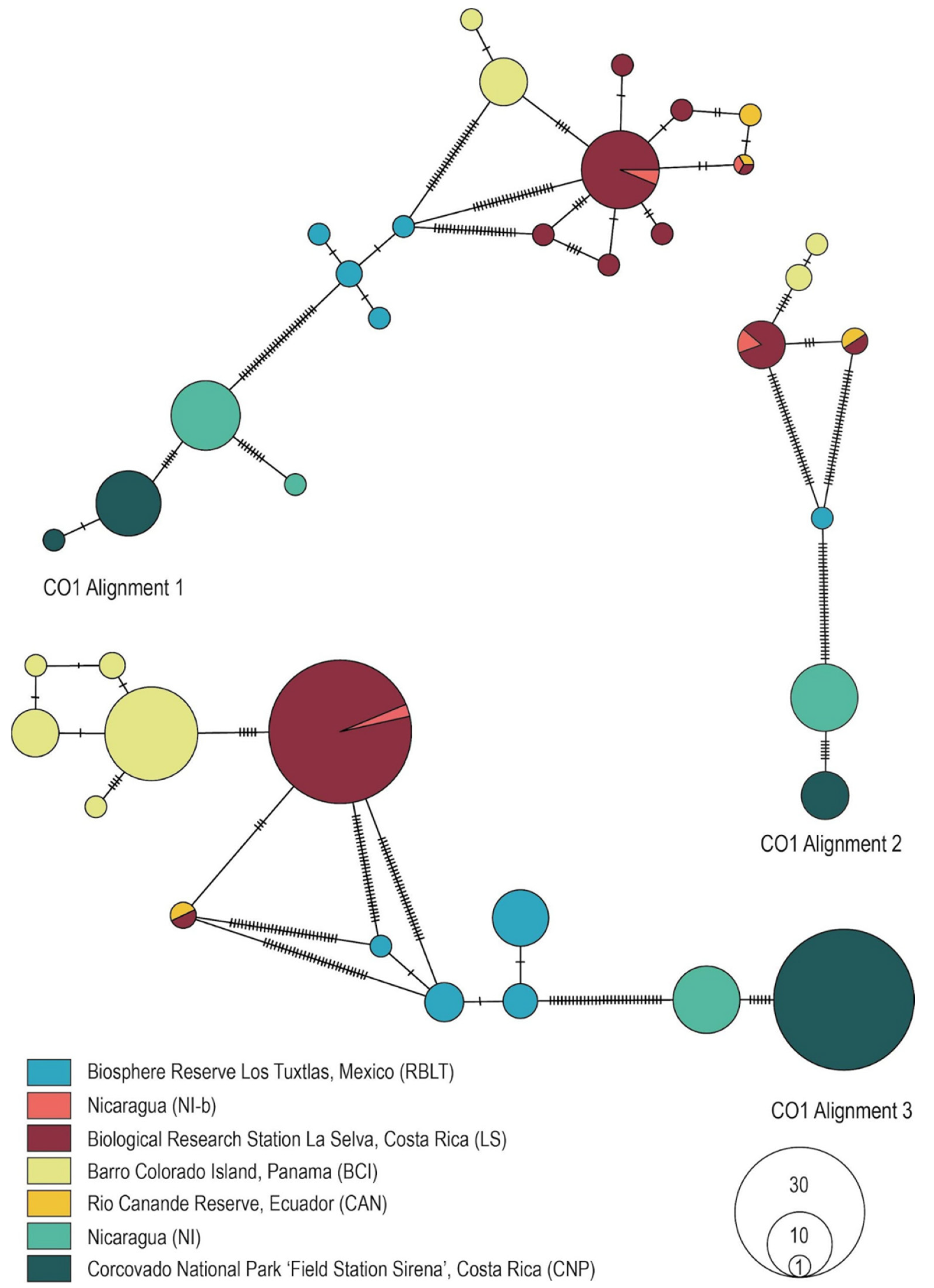
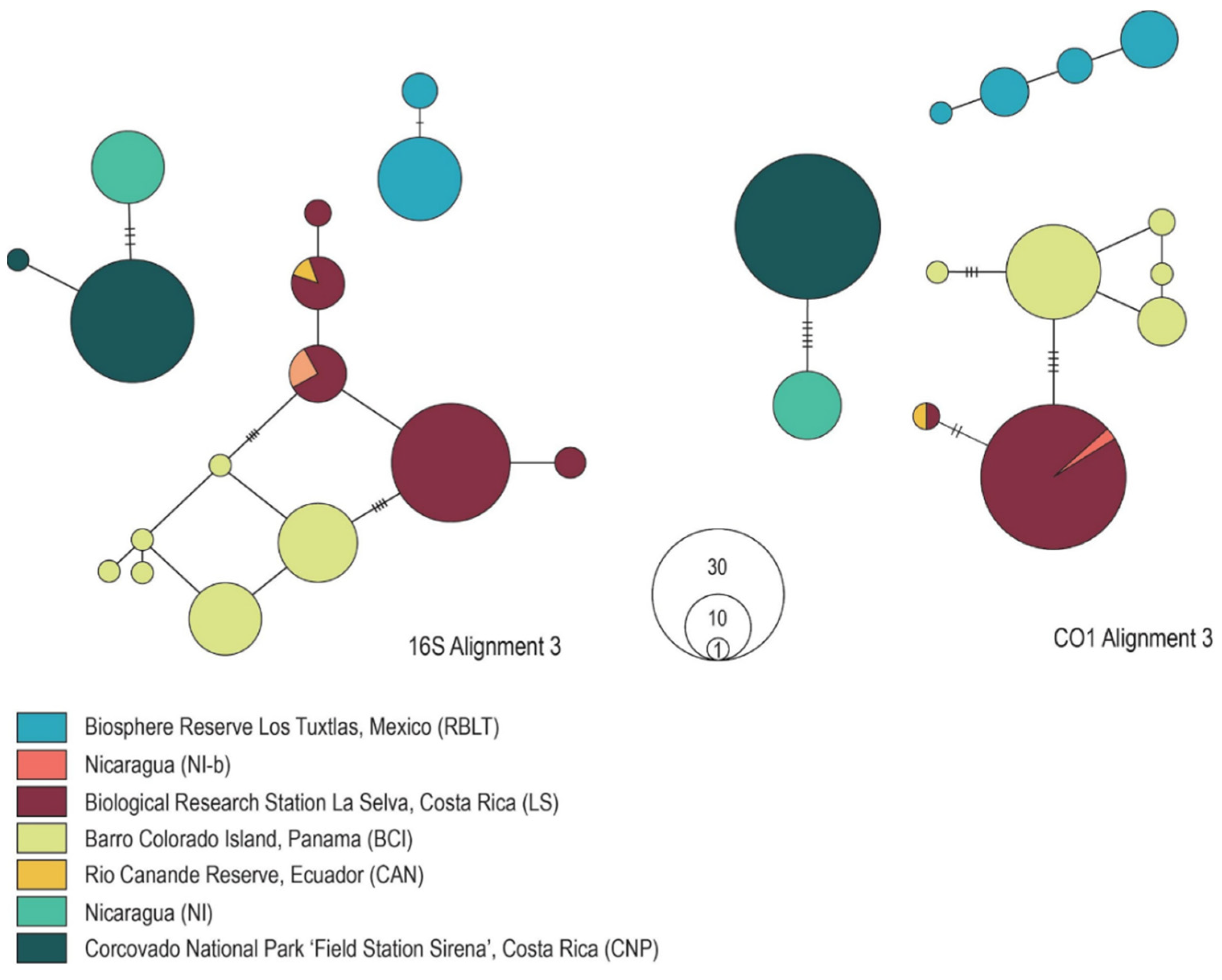
3.3. Genealogical Network Analyses
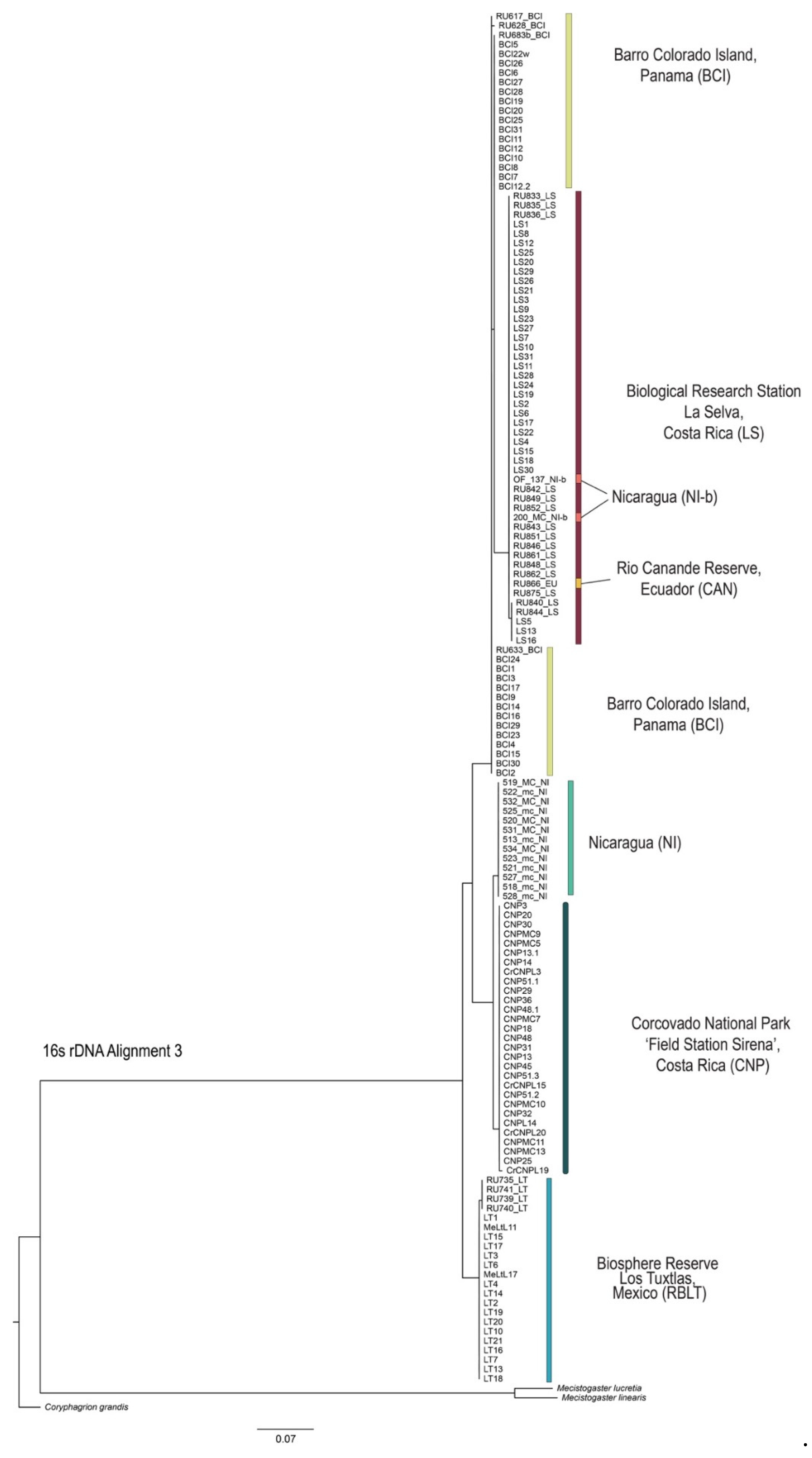
3.4. Phylogenetic Tree Reconstruction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.C.; Chen, M.Y.; Wang, J.F.; Liang, A.P.; Lin, C.P. Some mitochondrial genes perform better for damselfly phylogenetics: Species- and population-level analyses of four complete mitogenomes of Euphaea sibling species. Syst. Entomol. 2018, 43, 702–715. [Google Scholar] [CrossRef]
- Damm, S.; Schierwater, B.; Hadrys, H. An integrative approach to species discovery in odonates: From character-based DNA barcoding to ecology. Mol. Ecol. 2010, 19, 3881–3893. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.H.; Ron, S.R.; Santos, J.C.; Schneider, C.J.; Moritz, C. Integrating phylogenetics and environmental niche models to explore speciation mechanisms in dendrobatid frogs. Evolution 2004, 58, 1781–1793. [Google Scholar] [CrossRef]
- Paknia, O.; Bergmann, T.; Hadrys, H. Some ‘ant’swers: Application of a layered barcode approach to problems in ant taxonomy. Mol. Ecol. Resour. 2015, 15, 1262–1274. [Google Scholar] [CrossRef]
- Rach, J.; Bergmann, T.; Paknia, O.; DeSalle, R.; Schierwater, B.; Hadrys, H. The marker choice: Unexpected resolving power of an unexplored CO1 region for layered DNA barcoding approaches. PLoS ONE 2017, 12, e0174842. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, A.R.; Brown, J.M.; Stanger-Hall, K.; Lemmon, E.M. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst. Biol. 2009, 58, 130–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, T.H.; Rosenberg, M.S. Multiple sequence alignment accuracy and phylogenetic inference. Syst. Biol. 2006, 55, 314–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, S.; Blanke, A.; Meusemann, K. Reanalyzing the Palaeoptera problem—The origin of insect flight remains obscure. Arthropod Struct. Dev. 2018, 47, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Fincke, O.M. Use of forest and tree species, and dispersal by giant damselflies (Pseudostigmatidae): Their prospects in fragmented forests. In Forest and Dragonflies, 4th WDA International Symposium of Odonatology; Pensoft: Sofia, Bulgaria, 2006; pp. 103–125. [Google Scholar]
- Selys, L.E.d. Synopsis des Agrionines. Première Légion—Pseudostigma. Bull. L’académie R. Sci. Lett. Beaux-Arts Belg. 1860, 2, 9–27. [Google Scholar]
- Selys, L.E.d. Révision du synopsis des Agrionines, premiére partie comprenant des légions Psuedostigma—Podagrion—Platycnemis et Protoneura. Mémoire Cour. Académie R. Belg. 1886, 38, 233. [Google Scholar]
- Feindt, W.; Fincke, O.; Hadrys, H. Still a one species genus? Strong genetic diversification in the world’s largest living odonate, the Neotropical damselfly Megaloprepus caerulatus. Conserv. Genet. 2014, 15, 469–481. [Google Scholar] [CrossRef]
- Ris, F. Libellen (Odonata) aus der Region der amerikanischen Kordilleren von Costarica bis Catamarca. Arch. Nat. 1916, 82A, 1–197. [Google Scholar]
- Schmidt, E. Odonata nebst Bemerkungen über die Anomisma und Chalcopteryx des Amazonas-Gebiets. In 1941–1942 Beiträge zur Fauna Perus Nach der Ausbeute der Hamburger Südperu Expedition 1936; G. Fischer: Hamburg, German, 1942; Volume 2, pp. 225–276. [Google Scholar]
- Fincke, O.M.; Xu, M.; Khazan, E.S.; Wilson, M.; Ware, J.L. Tests of hypotheses for morphological and genetic divergence in Megaloprepus damselflies across Neotropical forests. Biol. J. Linn. Soc. 2018, 125, 844–861. [Google Scholar] [CrossRef]
- Damm, S.; Hadrys, H. Trithemis morrisoni sp. nov. and T. palustris sp. nov. from the Okavango and Upper Zambezi Floodplains previously hidden under T. stictica (Odonata: Libellulidae). Int. J. Odonatol. 2009, 12, 131–145. [Google Scholar] [CrossRef]
- De Mandal, S.; Chhakchhuak, L.; Gurusubramanian, G.; Kumar, N.S. Mitochondrial markers for identification and phylogenetic studies in insects—A Review. DNA Barcodes 2014, 2, 1–9. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the insect mitochondrial molecular clock: The mid-Aegean trench calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, V.; Schaeffer, B.; Missoup, A.D.; Kennis, J.; Colyn, M.; Denys, C.; Tatard, C.; Cruaud, C.; Laredo, C. Assessment of three mitochondrial genes (16S, Cytb, CO1) for identifying species in the Praomyini tribe (Rodentia: Muridae). PLoS ONE 2012, 7, e36586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Vogler, A.P.; Barraclough, T.G. Ecology has contrasting effects on genetic variation within species versus rates of molecular evolution across species in water beetles. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142476. [Google Scholar] [CrossRef] [Green Version]
- Shearer, T.; Van Oppen, M.; Romano, S.; Wörheide, G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 2002, 11, 2475–2487. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Ware, J.; May, M.; Kjer, K. Phylogeny of the higher Libelluloidea (Anisoptera: Odonata): An exploration of the most speciose superfamily of dragonflies. Mol. Phylogenetics Evol. 2007, 45, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, T.; Rach, J.; Damm, S.; DeSalle, R.; Schierwater, B.; Hadrys, H. The potential of distance-based thresholds and character-based DNA barcoding for defining problematic taxonomic entities by CO1 and ND1. Mol. Ecol. Resour. 2013, 13, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feindt, W.; Osigus, H.-J.; Herzog, R.; Mason, C.E.; Hadrys, H. The complete mitochondrial genome of the neotropical helicopter damselfly Megaloprepus caerulatus (Odonata: Zygoptera) assembled from next generation sequencing data. Mitochondrial DNA Part B 2016, 1, 497–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swofford, D.L. PAUP * Phylogenetic Analysis Using Parsimony (* and Other Methods). 2002. Available online: http://www2.ib.unicamp.br/profs/sfreis/SistematicaMolecular/Aula09MetodoParcimonia/Leituras/ThePhylogeneticHandbookParcimonia.pdf (accessed on 27 September 2022).
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution and the Genetics of Populations, Volume 2, The Theory of Gene Frequencies; University of Chicago Press: Chicago, IL, USA, 1969. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, L.F.; Clausnitzer, V.; Hadrys, H. Convergent evolution of gigantism in damselflies of Africa and South America? Evidence from nuclear and mitochondrial sequence data. Mol. Phylogenetics Evol. 2007, 42, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, E.F.; Bybee, S.M.; Erickson, R.J.; Condamine, F.L. Forest giants on different evolutionary branches: Ecomorphological convergence in helicopter damselflies. Evolution 2019, 73, 1045–1054. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In Selected Papers of Hirotugu Akaike; Springer: Berlin/Heidelberg, Germany, 1998; pp. 199–213. [Google Scholar]
- Tautz, D.; Arctander, P.; Minelli, A.; Thomas, R.H.; Vogler, A.P. A plea for DNA taxonomy. Trends Ecol. Evol. 2003, 18, 70–74. [Google Scholar] [CrossRef]
- Fincke, O.M.; Hadrys, H. Unpredictable offspring survivorship in the damselfly, Megaloprepus coerulatus, shapes parental behavior, constrains sexual selection, and challenges traditional fitness estimates. Evolution 2001, 55, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Hadrys, H.; Wargel, A.; Giere, S.; Kraus, B.; Streit, B. A panel of microsatellite markers to detect and monitor demographic bottlenecks in the riverine dragonfly Orthetrum coerulescens F. Mol. Ecol. Notes 2007, 7, 287–289. [Google Scholar] [CrossRef]
- Hadrys, H.; Timm, J.; Streit, B.; Giere, S. A panel of microsatellite markers to study sperm precedence patterns in the emperor dragonfly Anax imperator (Odonata: Anisoptera). Mol. Ecol. Notes 2007, 7, 296–298. [Google Scholar] [CrossRef]
- DeSalle, R.; Schierwater, B.; Hadrys, H. MtDNA: The small workhorse of evolutionary studies. Front. Biosci.-Landmark 2017, 22, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koroiva, R.; Pepinelli, M.; Rodrigues, M.E.; de Oliveira Roque, F.; Lorenz-Lemke, A.P.; Kvist, S. DNA barcoding of odonates from the Upper Plata basin: Database creation and genetic diversity estimation. PLoS ONE 2017, 12, e0182283. [Google Scholar] [CrossRef] [Green Version]
- Koroiva, R.; Kvist, S. Estimating the barcoding gap in a global dataset of cox1 sequences for Odonata: Close, but no cigar. Mitochondrial DNA Part A 2018, 29, 765–771. [Google Scholar] [CrossRef]
- Vega-Sánchez, Y.M.; Lorenzo-Carballa, M.O.; Vilela, D.S.; Guillermo-Ferreira, R.; Koroiva, R. Comment on “Molecular identification of seven new Zygopteran genera from South China through partial cytochrome oxidase subunit I (COI) gene”. Meta Gene 2020, 25, 100759. [Google Scholar] [CrossRef]
- Lorenzo-Carballa, M.O.; Sanmartín-Villar, I.; Cordero-Rivera, A. Molecular and Morphological Analyses Support Different Taxonomic Units for Asian and Australo-Pacific Forms of Ischnura aurora (Odonata, Coenagrionidae). Diversity 2022, 14, 606. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Damaso, N.; Robertson, J.M. Assessment of BOLD and GenBank—Their accuracy and reliability for the identification of biological materials. PLoS ONE 2019, 14, e0217084. [Google Scholar] [CrossRef] [Green Version]
- Arabi, J.; Cruaud, C.; Couloux, A.; Hassanin, A. Studying sources of incongruence in arthropod molecular phylogenies: Sea spiders (Pycnogonida) as a case study. Comptes Rendus Biol. 2010, 333, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef]
- Wilson, C.G.; Nowell, R.W.; Barraclough, T.G. Cross-contamination explains “inter and intraspecific horizontal genetic transfers” between asexual bdelloid rotifers. Curr. Biol. 2018, 28, 2436–2444.e2414. [Google Scholar] [CrossRef] [Green Version]
- Buhay, J.E. “COI-like” sequences are becoming problematic in molecular systematic and DNA barcoding studies. J. Crustac. Biol. 2009, 29, 96–110. [Google Scholar] [CrossRef]
- Feindt, W.; Hadrys, H. The damselfly genus Megaloprepus (Odonata: Pseudostigmatidae): Revalidation and delimitation of species-level taxa including the description of one new species. Zootaxa 2022, 5115, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Khazan, E.S. Tests of biological corridor efficacy for conservation of a Neotropical giant damselfly. Biol. Conserv. 2014, 177, 117–125. [Google Scholar] [CrossRef]
- Hansen, M.C.; Wang, L.; Song, X.-P.; Tyukavina, A.; Turubanova, S.; Potapov, P.V.; Stehman, S.V. The fate of tropical forest fragments. Sci. Adv. 2020, 6, eaax8574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brower, A.V.; Desalle, R. Practical and theoretical considerations for choice of a DNA sequence region in insect molecular systematics, with a short review of published studies using nuclear gene regions. Ann. Entomol. Soc. Am. 1994, 87, 702–716. [Google Scholar] [CrossRef]
- Damm, S.; Dijkstra, K.-D.B.; Hadrys, H. Red drifters and dark residents: The phylogeny and ecology of a Plio-Pleistocene dragonfly radiation reflects Africa’s changing environment (Odonata, Libellulidae, Trithemis). Mol. Phylogenetics Evol. 2010, 54, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Damm, S.; Hadrys, H. A dragonfly in the desert: Genetic pathways of the widespread Trithemis arteriosa (Odonata: Libellulidae) suggest male-biased dispersal. Org. Divers. Evol. 2012, 12, 267–279. [Google Scholar] [CrossRef]
- Dijkstra, K.-D.B.; Kalkman, V.J.; Dow, R.A.; Stokvis, F.R.; Van Tol, J. Redefining the damselfly families: A comprehensive molecular phylogeny of Zygoptera (Odonata). Syst. Entomol. 2014, 39, 68–96. [Google Scholar] [CrossRef]
- Vega-Sánchez, Y.M.; Mendoza-Cuenca, L.F.; González-Rodríguez, A. Complex evolutionary history of the American Rubyspot damselfly, Hetaerina americana (Odonata): Evidence of cryptic speciation. Mol. Phylogenetics Evol. 2019, 139, 106536. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Anderson, C.; Buckley, T.; Erpenbeck, D.; Rickert, A.; Misof, K. An empirical analysis of mt 16S rRNA covarion-like evolution in insects: Site-specific rate variation is clustered and frequently detected. J. Mol. Evol. 2002, 55, 460–469. [Google Scholar] [CrossRef]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.V.; Yuhki, N.; Masuda, R.; Modi, W.; O’Brien, S.J. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 1994, 39, 174–190. [Google Scholar] [CrossRef]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 2008, 105, 13486–13491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ožana, S.; Dolný, A.; Pánek, T. Nuclear copies of mitochondrial DNA as a potential problem for phylogenetic and population genetic studies of Odonata. Syst. Entomol. 2022, 47, 591–602. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org/). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Location | CO1 | 16S rDNA | ||
|---|---|---|---|---|
| N Study-A | N Study-B | N Study-A | N Study-B | |
| Biosphere Reserve Los Tuxtlas, Mexico (RBLT) | 1 | 18 | 4 | 18 |
| Cusuco National Park, Honduras (HN) | / | / | / | / |
| Nicaragua * (NI) | 11 | / | 13 | / |
| Nicaragua * (NI-b) | 1 | / | 2 | / |
| Biological Research Station La Selva, Costa Rica (LS) | 6 | 30 | 15 | 30 |
| Corcovado National Park ‘Field Station Sirena’, Costa Rica (CNP) | 6 | 29 | / | 29 |
| Barro Colorado Island, Panama (BCI) | 3 | 29 | 4 | 29 |
| Rio Canande Reserve, Ecuador (CAN) | 1 | / | 1 | / |
| N total | 29 | 106 | 39 | 106 |
| 16S rDNA | ||||||
|---|---|---|---|---|---|---|
| Population * | N | S | H | Hd (±SD) | π (±SD) | |
| Alignment 1 577 bp | BCI | 7 | 15 | 4 | 0.714 (0.181) | 0.015 (0.007) |
| RBLT | 5 | 4 | 2 | 0.400 (0.237) | 0.006 (0.003) | |
| LS | 18 | 11 | 4 | 0.399 (0.138) | 0.003 (0.001) | |
| CAN | 2 | 0 | 2 | 0.00 | 0.00 | |
| HN | 1 | 0 | 1 | 0.00 | 0.00 | |
| NI | 14 | 3 | 3 | 0.385 (0.149) | 0.002 (0.003) | |
| NI-b | 2 | 3 | 2 | 1.000 (0.500) | 0.008 (0.006) | |
| Total | 49 | 29 | 10 | 0.759 (0.047) | 0.021 (0.003) | |
| Alignment 2 325 bp | BCI | 4 | 2 | 3 | 0.833 (0.222) | 0.003 (0.001) |
| RBLT | 4 | 0 | 1 | 0.00 | 0.00 | |
| LS | 15 | 1 | 2 | 0.248 (0.131) | 0.001 (0.000) | |
| CAN | 1 | 0 | 1 | 0.00 | 0.00 | |
| NI | 13 | 3 | 2 | 1.00 (0.250) | 0.008 (0.000) | |
| NI-b | 2 | 0 | 1 | 0.00 | 0.00 | |
| Total | 39 | 26 | 7 | 0.722 (0.047) | 0.029 (0.002) | |
| Alignment 3 321 bp | BCI | 33 | 2 | 3 | 0.544 (0.036) | 0.002 (0.000) |
| CNP | 29 | 1 | 2 | 0.069 (0.063) | 0.000 (0.000) | |
| RBLT | 22 | 1 | 2 | 0.312 (0.106) | 0.001 (0.000) | |
| LS | 45 | 1 | 2 | 0.202 (0.073) | 0.001 (0.000) | |
| CAN | 1 | 0 | 1 | 0.00 | 0.00 | |
| NI | 13 | 0 | 1 | 0.00 | 0.00 | |
| NI-b | 2 | 0 | 1 | 0.00 | 0.00 | |
| Total | 145 | 27 | 10 | 0.831 (0.015) | 0.029 (0.001) | |
| CO1 | ||||||
|---|---|---|---|---|---|---|
| Population * | N | S | H | Hd (±SD) | π (±SD) | |
| Alignment 1 ** 419 bp | BCI | 7 | / | / | / | / |
| CNP | 11 | / | / | / | / | |
| RBLT | 5 | / | / | / | / | |
| LS | 21 | / | / | / | / | |
| CAN | 2 | / | / | / | / | |
| NI | 13 | / | / | / | / | |
| NI-b | 2 | / | / | / | / | |
| Total | 61 | / | / | / | / | |
| Alignment 2 395 bp | BCI | 3 | 1 | 2 | 0.667 (0.314) | 0.002 (0.001) |
| CNP | 6 | 0 | 1 | 0.00 | 0.00 | |
| RBLT | 1 | 0 | 1 | 0.00 | 0.00 | |
| LS | 6 | 3 | 2 | 0.333 (0.215) | 0.003 (0.002) | |
| CAN | 1 | 0 | 1 | 0.00 | 0.00 | |
| NI | 11 | 0 | 1 | 0.00 | 0.00 | |
| NI-b | 1 | 0 | 1 | 0.00 | 0.00 | |
| Total | 29 | 54 | 9 | 0.786 (0.049) | 0.052 (0.004) | |
| Alignment 3 395 bp | BCI | 32 | 6 | 5 | 0.502 (0.093) | 0.002 (0.006) |
| CNP | 35 | 0 | 1 | 0.00 | 0.00 | |
| RBLT | 19 | 3 | 4 | 0.713 (0.058) | 0.003 (0.000) | |
| LS | 36 | 3 | 2 | 0.056 (0.052) | 0.000 (0.000) | |
| CAN | 1 | 0 | 1 | 0.00 | 0.00 | |
| NI | 11 | 0 | 1 | 0.00 | 0.00 | |
| NI-b | 1 | 0 | 1 | 0.00 | 0.00 | |
| Total | 135 | 59 | 15 | 0.826 (0.017) | 0.055 (0.002) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feindt, W.; Hadrys, H. The Quality of Sequence Data Affects Biodiversity and Conservation Perspectives in the Neotropical Damselfly Megaloprepus caerulatus. Diversity 2022, 14, 1056. https://doi.org/10.3390/d14121056
Feindt W, Hadrys H. The Quality of Sequence Data Affects Biodiversity and Conservation Perspectives in the Neotropical Damselfly Megaloprepus caerulatus. Diversity. 2022; 14(12):1056. https://doi.org/10.3390/d14121056
Chicago/Turabian StyleFeindt, Wiebke, and Heike Hadrys. 2022. "The Quality of Sequence Data Affects Biodiversity and Conservation Perspectives in the Neotropical Damselfly Megaloprepus caerulatus" Diversity 14, no. 12: 1056. https://doi.org/10.3390/d14121056