
Population Structure of White Sturgeon (Acipenser transmontanus) in the Columbia River Inferred from Single-Nucleotide Polymorphisms
Abstract
:1. Introduction
2. Methods
2.1. Individual-Based Analyses
2.2. Group-Based Analyses
2.3. Dispersal
3. Results
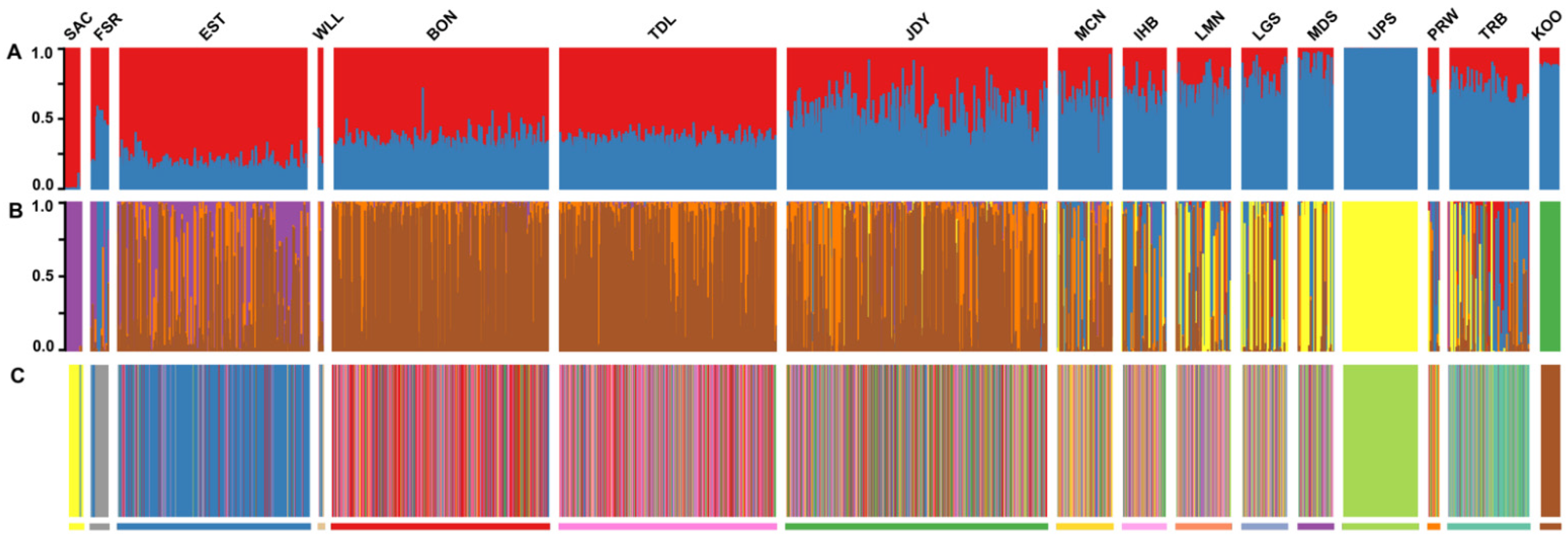
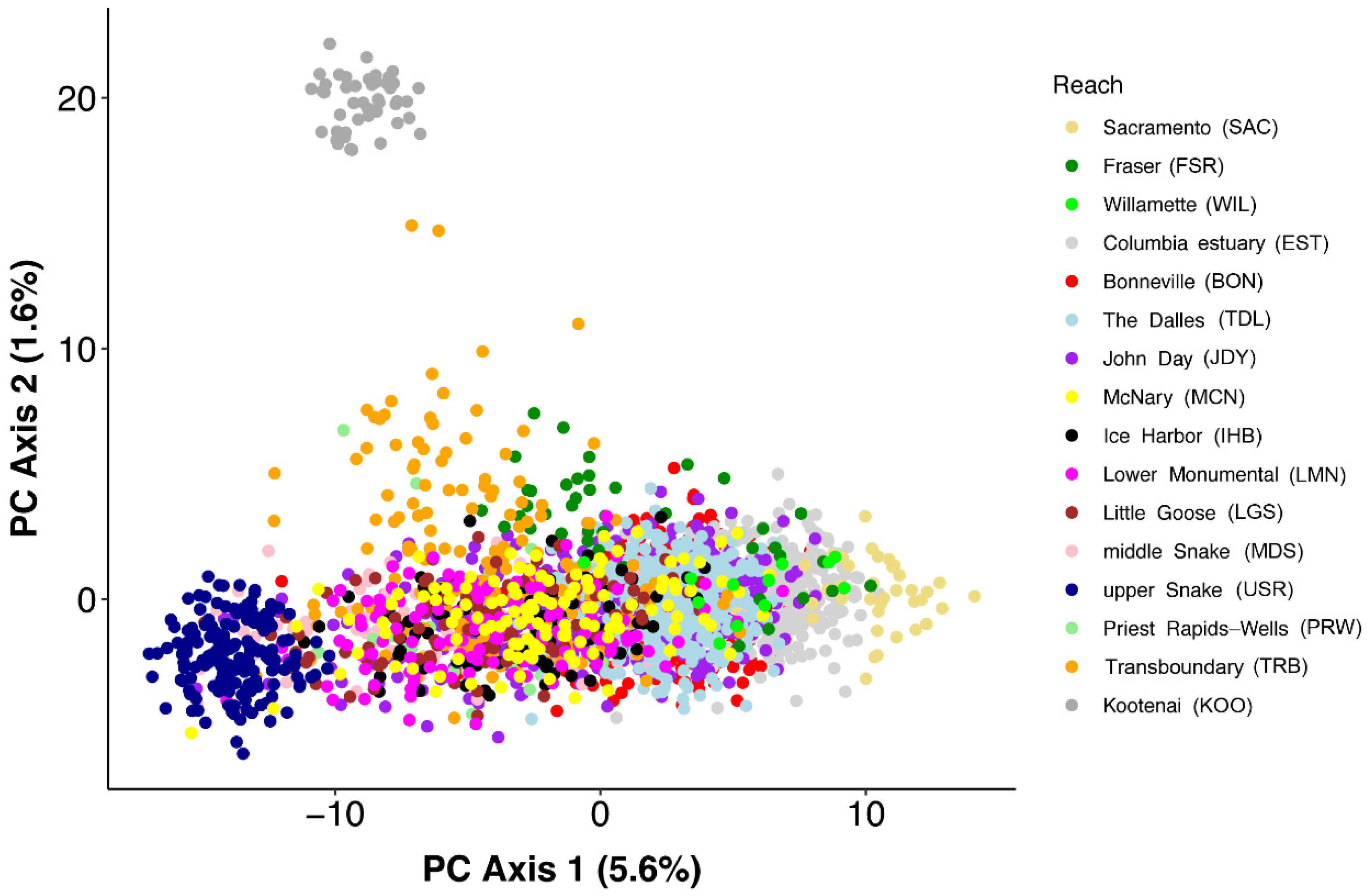
3.1. Individual-Based Analyses
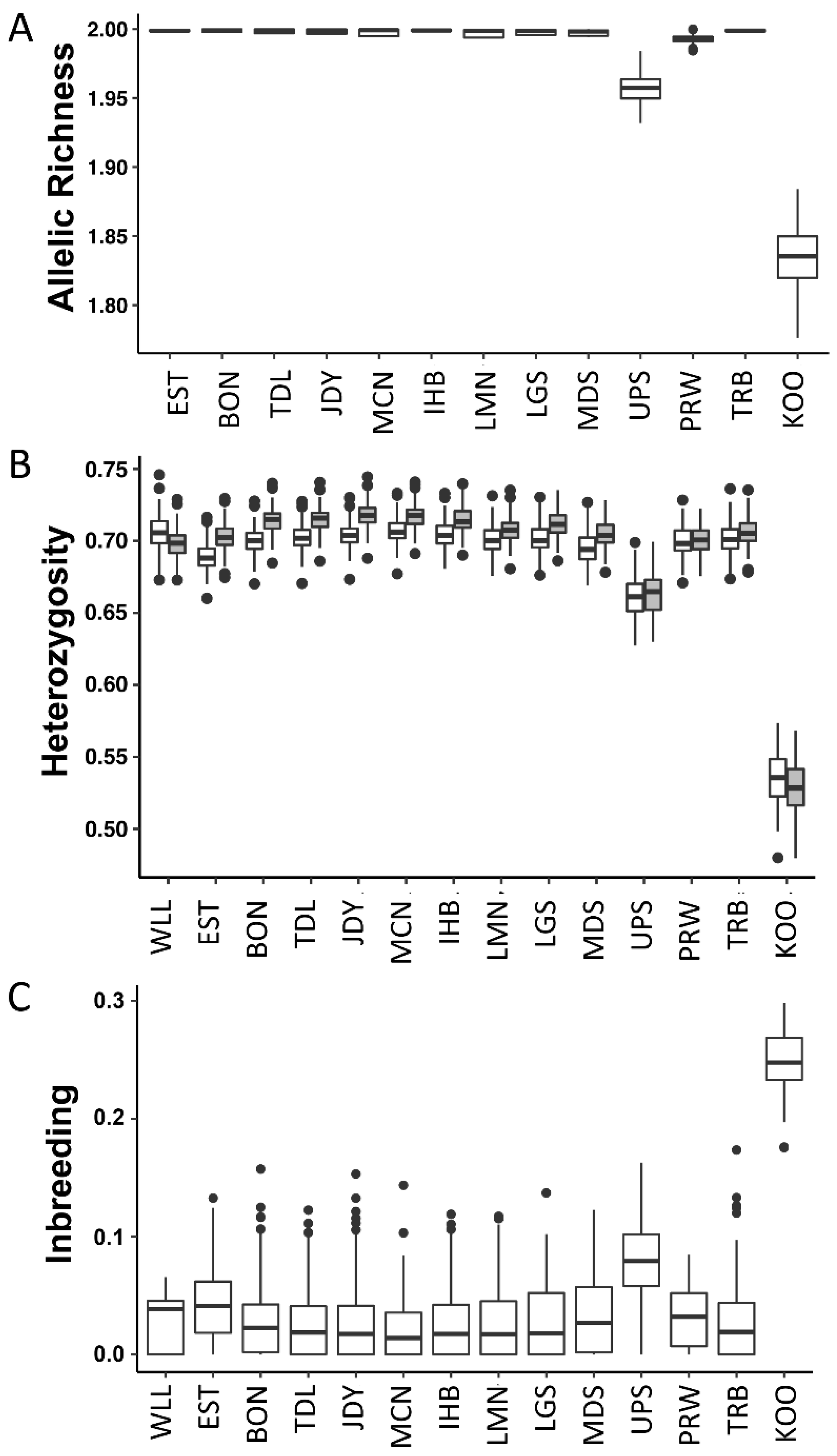
3.2. Group-Based Analyses
3.3. Identification of Dispersal
4. Discussion
4.1. Spatial Population Structure
4.2. Genetic Diversity
4.3. Dispersal
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rien, T.A.; Beamesderfer, R.C. Accuracy and Precision of White Sturgeon Age Estimates from Pectoral Fin Rays. Trans. Am. Fish. Soc. 1994, 123, 255–265. [Google Scholar] [CrossRef]
- Hildebrand, L.R.; Drauch Schreier, A.; Lepla, K.; McAdam, S.O.; McLellan, J.; Parsley, M.J.; Paragamian, V.L.; Young, S.P. Status of White Sturgeon (Acipenser transmontanus Richardson, 1863) throughout the species range, threats to survival, and prognosis for the future. J. Appl. Ichthyol. 2016, 32, 261–312. [Google Scholar] [CrossRef]
- Winemiller, K.O. Life history strategies, population regulation, and implications for fisheries management. Can. J. Fish. Aquat. Sci. 2005, 62, 872–885. [Google Scholar] [CrossRef]
- Galbreath, J.L. Status, life history, management of Columbia river white sturgeon, Acipenser transmontanus. Environ. Biol. Fishes 1985, 14. [Google Scholar]
- Doroshov, S.I.; Moberg, G.P.; van Eenennaam, J.P. Observations on the reproductive cycle of cultured white sturgeon, Acipenser transmontanus. Environ. Biol. Fishes 1997, 48, 265–278. [Google Scholar] [CrossRef]
- Parsley, M.J.; Beckman, L.G. White Sturgeon Spawning and Rearing Habitat in the Lower Columbia River. North Am. J. Fish. Manag. 1994, 14, 812–827. [Google Scholar] [CrossRef]
- Scott, W.; Crossman, E. Freshwater Fishes of Canada, Bulletin 184; Fisheries Research Board of Canada: Ottawa, Canada, 1973; p. 966. [Google Scholar]
- Lane, E.D. Status of the White Sturgeon, Acipenser-Transmontanus, in Canada. Can. Field-Nat. 1991, 105. [Google Scholar]
- McCabe, G.T.; Tracy, C.A. Spawning and early life history of white sturgeon, Acipenser transmontanus, in the lower Columbia River. Fish. Bull. 1994, 92, 760–772. [Google Scholar]
- Beamesderfer, R.C.P.; Rien, T.A.; Nigro, A.A. Differences in the Dynamics and Potential Production of Impounded and Unimpounded White Sturgeon Populations in the Lower Columbia River. Trans. Am. Fish. Soc. 1995, 124, 857–872. [Google Scholar] [CrossRef]
- Parsley, M.J.; Kappenman, K.M. White sturgeon spawning areas in the lower Snake River. Northwest Sci. 2000, 74, 192–201. [Google Scholar]
- Counihan, T.D.; Miller, A.I.; Parsley, M.J. Indexing the Relative Abundance of Age-0 White Sturgeons in an Impoundment of the Lower Columbia River from Highly Skewed Trawling Data. North Am. J. Fish. Manag. 1999, 19, 520–529. [Google Scholar] [CrossRef]
- Schreier, A.; Stephenson, S.; Rust, P.; Young, S. The case of the endangered Kootenai River white sturgeon (Acipenser transmontanus) highlights the importance of post-release genetic monitoring in captive and supportive breeding programs. Biol. Conserv. 2015, 192, 74–81. [Google Scholar] [CrossRef]
- Thorstensen, M.; Bates, P.; Lepla, K.; Schreier, A. To breed or not to breed? Maintaining genetic diversity in white sturgeon supplementation programs. Conserv. Genet. 2019, 20, 997–1007. [Google Scholar] [CrossRef]
- Schreier, A.D.; Rodzen, J.; Ireland, S.; May, B. Genetic techniques inform conservation aquaculture of the endangered Kootenai river white sturgeon Acipenser transmontanus. Endanger. Species Res. 2012, 16, 65–75. [Google Scholar] [CrossRef]
- Wood, C.C.; Sneep, D.; McAdam, S.; Korman, J.; Hatfield, T. Recovery Potential Assessment for White Sturgeon Populations Listed under the Species at Risk Act; Canadian Science Advisory Secretariat: Ottawa, Canada, 2007. [Google Scholar]
- Schreier, A.D.; Mahardja, B.; May, B. Patterns of population structure vary across the range of the white sturgeon. Trans. Am. Fish. Soc. 2013, 142, 1273–1286. [Google Scholar] [CrossRef]
- Campbell, N.R.; Harmon, S.A.; Narum, S.R. Genotyping-in-Thousands by sequencing (GT-seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 2015, 15, 855–867. [Google Scholar] [CrossRef]
- Delomas, T.A.; Willis, S.C.; Parker, B.L.; Miller, D.; Anders, P.; Schreier, A.; Narum, S. Genotyping single nucleotide polymorphisms and inferring ploidy by amplicon sequencing for polyploid, ploidy-variable organisms. Mol. Ecol. Resour. 2021, 21, 2288–2298. [Google Scholar] [CrossRef]
- Schreier, A.D.; Gille, D.; Mahardja, B.; May, B. Neutral markers confirm the octoploid origin and reveal spontaneous autopolyploidy in white sturgeon, Acipenser transmontanus. J. Appl. Ichthyol. 2011, 27, 24–33. [Google Scholar] [CrossRef]
- Huang, K.; Guo, S.T.; Shattuck, M.R.; Chen, S.T.; Qi, X.G.; Zhang, P.; Li, B.G. A maximum-likelihood estimation of pairwise relatedness for autopolyploids. Heredity 2015, 114, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Dunn, D.W.; Ritland, K.; Li, B. Polygene: Population genetics analyses for autopolyploids based on allelic phenotypes. Methods Ecol. E 2020, 11, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S.; Anderson, E.C. Purging putative siblings from population genetic data sets: A cautionary view. Mol. Ecol. 2017, 26, 1211–1224. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Effects of sampling close relatives on some elementary population genetics analyses. Mol. Ecol. Resour. 2018, 18, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software Structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Grubbs, F.E. Procedures for Detecting Outlying Observations in Samples. Technometrics 1969, 11, 1–21. [Google Scholar] [CrossRef]
- Willis, S.C.; Macrander, J.; Farias, I.P.; Orti, G. Simultaneous delimitation of species and quantification of interspecific hybridization in Amazonian peacock cichlids (genus Cichla) using multi-locus data. BMC EBiol. 2012, 12, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paetkau, D.; Slade, R.; Burdens, M.; Estoup, A. Genetic assignment methods for the direct, real-time estimation of migration rate: A simulation-based exploration of accuracy and power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G. Genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 2020, 20, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Meirmans, P.G.; Liu, S.; van Tienderen, P.H. The Analysis of Polyploid Genetic Data. J. Hered. 2018, 109, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Adamack, A.T.; Gruber, B. PopGenReport: Simplifying basic population genetic analyses in R. Methods Ecol. E 2014, 5, 384–387. [Google Scholar] [CrossRef]
- Jones, O.R.; Wang, J. COLONY: A program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 2010, 10, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Rodzen, J.A.; Famula, T.R.; May, B. Estimation of parentage and relatedness in the polyploid white sturgeon (Acipenser transmontanus) using a dominant marker approach for duplicated microsatellite loci. Aquaculture 2004, 232, 165–182. [Google Scholar] [CrossRef]
- Waples, R.S.; Teel, D.J.; Myers, J.M.; Marshall, A.R. Life-history divergence in Chinook salmon: Historic contingency and parallel evolution. Evolution 2004, 58, 386–403. [Google Scholar] [CrossRef] [Green Version]
- Narum, S.R.; Hess, J.E.; Matala, A.P. Examining Genetic Lineages of Chinook Salmon in the Columbia River Basin. Trans. Am. Fish. Soc. 2010, 139, 1465–1477. [Google Scholar] [CrossRef]
- Collins, E.; Hargrove, J.; Delomas, T.; Narum, S.R. Distribution of genetic variation underlying adult migration timing in steelhead of the Columbia River basin. Ecol. E 2020, in press. [Google Scholar] [CrossRef]
- Parsley, M.J.; Wright, C.D.; van der Leeuw, B.K.; Kofoot, E.E.; Peery, C.A.; Moser, M.L. White sturgeon (Acipenser transmontanus) passage at the Dalles Dam, Columbia River, USA. J. Appl. Ichthyol. 2007, 23, 627–635. [Google Scholar] [CrossRef]
- Moody, M.E.; Mueller, L.D.; Soltis, D.E. Genetic variation and random drift in autotetraploid populations. Genetics 1993, 134, 649–657. [Google Scholar] [CrossRef]
- Parsley, M.J.; Anders, P.J.; Miller, A.I.; Beckman, L.G.; McCabe, G.T. Recovery of white sturgeon populations through natural production: Understanding the influence of abiotic and biotic factors on spawning and subsequent recruitment. Am. Fish. Soc. Symp. 2002, 2002, 55–66. [Google Scholar]
- Rieman, B.E.; Beamesderfer, R.C. White Sturgeon in the Lower Columbia River: Is the Stock Overexploited. North Am. J. Fish. Manag. 1990, 10, 388–396. [Google Scholar] [CrossRef]
- Parker, B.L.; Beamesderfer, R.C.; Powell, J.; Orton, E.; Miller, D. White Sturgeon Hatchery Step I Master Plan for Lower Columbia and Snake River Impoundments; U.S. Department of Energy Office of Scientific and Technical Information: Portland, OR, USA, 2015.
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. E 2012, 27, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, P.W.; Adams, J.R.; Vucetich, J.A. Reevaluating and Broadening the Definition of Genetic Rescue. Conserv. Biol. 2011, 25, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Visger, C.J.; Marchant, D.B.; Soltis, P.S. Polyploidy: Pitfalls and paths to a paradigm. Am. J. Bot. 2016, 103, 1146–1166. [Google Scholar] [CrossRef] [PubMed]
- Idaho Power Company. Snake River White Sturgeon Conservation Plan, 2015–2020 Planning and Implementation; Idaho Power Company: Boise, ID, USA, 2015. [Google Scholar]
- Chung, Y.; Hey, J. Bayesian analysis of evolutionary divergence with genomic data under diverse demographic models. Mol. Biol. E 2017, 34, 1517–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaug, H.J. Allele-sharing methods for estimation of population size. Biometrics 2001, 57, 750–756. [Google Scholar] [CrossRef]
- Ruzzante, D.E.; McCracken, G.R.; Førland, B.; MacMillan, J.; Notte, D.; Buhariwalla, C.; Skaug, H.; Flemming, J.M. Validation of close-kin mark–recapture (CKMR) methods for estimating population abundance. Methods Ecol. E 2019, 10, 1445–1453. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reach | Code | N | r |
|---|---|---|---|
| Sacramento-San Joaquin | SAC | 37 | 37 |
| Fraser River | FSR | 44 | 42 |
| Columbia estuary * | EST | 490 | 432 |
| Willamette | WLL | 13 | 13 |
| Bonneville | BON | 504 | 494 |
| The Dalles | TDL | 505 | 499 |
| John Day | JDY | 669 | 599 |
| McNary | MCN | 178 | 125 |
| Ice Harbor | IHB | 108 | 101 |
| Lower Monumental | LMN | 132 | 124 |
| Little Goose | LGS | 112 | 106 |
| middle Snake | MDS | 90 | 82 |
| upper Snake | UPS | 264 | 169 |
| Priest Rapids-Wells Tailrace | PRW | 48 | 26 |
| Transboundary | TRB | 225 | 183 |
| Kootenai River | KOO | 49 | 46 |
| Assignment (Percentage) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAC | FSR | EST | WLL | BON | TDL | JDY | MCN | IHB | LMN | LGS | MDS | UPS | PRW | TRB | KOO | ||||
| Source | Sacramento | SAC | 37 | 91.9 | 0.0 | 8.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Fraser | FSR | 42 | 0.0 | 69.0 | 28.6 | 2.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| Columbia estuary | EST | 432 | 0.2 | 0.0 | 82.9 | 1.4 | 7.2 | 6.7 | 1.4 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| Willamette | WLL | 13 | 0.0 | 0.0 | 61.5 | 23.1 | 0.0 | 7.7 | 7.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| Bonneville | BON | 494 | 0.0 | 0.0 | 12.3 | 0.8 | 41.9 | 26.5 | 10.9 | 4.5 | 1.2 | 0.4 | 0.6 | 0.6 | 0.0 | 0.0 | 0.2 | 0.0 | |
| The Dalles | TDL | 499 | 0.0 | 0.0 | 10.0 | 0.4 | 25.5 | 42.9 | 16.0 | 2.8 | 0.8 | 1.0 | 0.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| John Day | JDY | 599 | 0.0 | 0.0 | 9.5 | 0.7 | 16.5 | 18.2 | 25.9 | 7.0 | 3.0 | 3.2 | 5.7 | 6.8 | 1.2 | 0.7 | 1.7 | 0.0 | |
| McNary | MCN | 125 | 0.0 | 0.0 | 3.2 | 0.0 | 8.0 | 7.2 | 12.8 | 25.6 | 7.2 | 11.2 | 10.4 | 8.8 | 3.2 | 0.8 | 1.6 | 0.0 | |
| Ice Harbor | IHB | 101 | 0.0 | 0.0 | 1.0 | 1.0 | 2.0 | 3.0 | 14.9 | 7.9 | 22.8 | 15.8 | 8.9 | 14.9 | 4.0 | 2.0 | 2.0 | 0.0 | |
| Lower Monumental | LMN | 124 | 0.0 | 0.0 | 1.6 | 0.0 | 2.4 | 3.2 | 11.3 | 8.1 | 10.5 | 29.0 | 9.7 | 17.7 | 4.0 | 0.8 | 1.6 | 0.0 | |
| Little Goose | LGS | 106 | 0.0 | 0.0 | 0.0 | 0.0 | 3.8 | 6.6 | 5.7 | 17.9 | 5.7 | 7.5 | 19.8 | 17.9 | 9.4 | 1.9 | 3.8 | 0.0 | |
| middle Snake | MDS | 82 | 0.0 | 0.0 | 0.0 | 0.0 | 1.2 | 1.2 | 14.6 | 4.9 | 8.5 | 11.0 | 9.8 | 28.0 | 19.5 | 1.2 | 0.0 | 0.0 | |
| upper Snake | UPS | 169 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100.0 | 0.0 | 0.0 | 0.0 | |
| Priest Rapids-Wells | PRW | 26 | 0.0 | 0.0 | 0.0 | 0.0 | 3.8 | 0.0 | 11.5 | 11.5 | 3.8 | 7.7 | 0.0 | 19.2 | 0.0 | 30.8 | 11.5 | 0.0 | |
| Transboundary | TRB | 183 | 0.0 | 0.0 | 0.5 | 0.0 | 0.5 | 2.7 | 7.1 | 4.9 | 3.8 | 2.7 | 1.6 | 4.9 | 3.8 | 1.1 | 65.0 | 1.1 | |
| Kootenai | KOO | 46 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100.0 | |
| SAC | FSR | EST | WLL | BON | TDL | JDY | MCN | IHB | LMN | LGS | MDS | UPS | PRW | TRB | KOO | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sacramento | SAC | -- | 0.073 | 0.022 | 0.023 | 0.043 | 0.045 | 0.060 | 0.083 | 0.097 | 0.106 | 0.107 | 0.129 | 0.229 | 0.109 | 0.110 | 0.289 |
| Fraser | FSR | 0.073 | -- | 0.034 | 0.034 | 0.029 | 0.029 | 0.029 | 0.036 | 0.042 | 0.047 | 0.045 | 0.055 | 0.123 | 0.043 | 0.045 | 0.180 |
| Columbia estuary | EST | 0.022 | 0.034 | -- | 0.002 | 0.007 | 0.008 | 0.016 | 0.031 | 0.041 | 0.048 | 0.049 | 0.066 | 0.153 | 0.050 | 0.050 | 0.227 |
| Willamette | WLL | 0.023 | 0.034 | 0.002 | -- | 0.007 | 0.009 | 0.014 | 0.028 | 0.036 | 0.046 | 0.046 | 0.063 | 0.148 | 0.046 | 0.047 | 0.224 |
| Bonneville | BON | 0.043 | 0.029 | 0.007 | 0.007 | -- | 0.001 | 0.003 | 0.012 | 0.018 | 0.023 | 0.023 | 0.035 | 0.109 | 0.025 | 0.027 | 0.202 |
| The Dalles | TDL | 0.045 | 0.029 | 0.008 | 0.009 | 0.001 | -- | 0.003 | 0.010 | 0.017 | 0.022 | 0.022 | 0.034 | 0.106 | 0.026 | 0.026 | 0.200 |
| John Day | JDY | 0.060 | 0.029 | 0.016 | 0.014 | 0.003 | 0.003 | -- | 0.004 | 0.008 | 0.012 | 0.012 | 0.020 | 0.082 | 0.013 | 0.016 | 0.186 |
| McNary | MCN | 0.083 | 0.036 | 0.031 | 0.028 | 0.012 | 0.010 | 0.004 | -- | 0.002 | 0.004 | 0.002 | 0.009 | 0.058 | 0.008 | 0.010 | 0.175 |
| Ice Harbor | IHB | 0.097 | 0.042 | 0.041 | 0.036 | 0.018 | 0.017 | 0.008 | 0.002 | -- | -- | -- | 0.005 | 0.049 | 0.007 | 0.009 | 0.177 |
| Lower Monumental | LMN | 0.106 | 0.047 | 0.048 | 0.046 | 0.023 | 0.022 | 0.012 | 0.004 | -- | -- | -- | 0.003 | 0.042 | 0.006 | 0.010 | 0.175 |
| Little Goose | LGS | 0.107 | 0.045 | 0.049 | 0.046 | 0.023 | 0.022 | 0.012 | 0.002 | -- | -- | -- | 0.002 | 0.041 | 0.006 | 0.009 | 0.169 |
| middle Snake | MDS | 0.129 | 0.055 | 0.066 | 0.063 | 0.035 | 0.034 | 0.020 | 0.009 | 0.005 | 0.003 | 0.002 | -- | 0.026 | 0.005 | 0.011 | 0.170 |
| upper Snake | UPS | 0.229 | 0.123 | 0.153 | 0.148 | 0.109 | 0.106 | 0.082 | 0.058 | 0.049 | 0.042 | 0.041 | 0.026 | -- | 0.040 | 0.048 | 0.189 |
| Priest Rapids-Wells | PRW | 0.109 | 0.043 | 0.050 | 0.046 | 0.025 | 0.026 | 0.013 | 0.008 | 0.007 | 0.006 | 0.006 | 0.005 | 0.040 | -- | 0.007 | 0.173 |
| Transboundary | TRB | 0.110 | 0.045 | 0.050 | 0.047 | 0.027 | 0.026 | 0.016 | 0.010 | 0.009 | 0.010 | 0.009 | 0.011 | 0.048 | 0.007 | -- | 0.147 |
| Kootenai | KOO | 0.289 | 0.180 | 0.227 | 0.224 | 0.202 | 0.200 | 0.186 | 0.175 | 0.177 | 0.175 | 0.169 | 0.170 | 0.189 | 0.173 | 0.147 | -- |
| Reaches | CKMR |
|---|---|
| Columbia estuary (EST) and Willamette (WIL) | 1 |
| Columbia estuary (EST) and The Dalles (TDL) | 1 |
| Bonneville (BON) and John Day (JDY) | 3 |
| John Day (JDY) and McNary (MCN) | 4 |
| John Day (JDY) and Ice Harbor (IHB) | 1 |
| John Day (JDY) and Lower Monumental (LMN) | 1 |
| John Day (JDY) and Priest Rapids-Wells Tailrace (PRW) | 3 |
| McNary (MCN) and Lower Monumental (LMN) | 1 |
| McNary (MCN) and Little Goose (LGS) | 3 |
| McNary (MCN) and Priest Rapids-Wells Tailrace (PRW) | 7 |
| Ice Harbor (IHB) and Lower Monumental (LMN) | 3 |
| Ice Harbor (IHB) and Little Goose (LGS) | 1 |
| Ice Harbor (IHB) and middle Snake (MDS) | 2 |
| Lower Monumental (LMN) and Little Goose (LGS) | 7 |
| Lower Monumental (LMN) and middle Snake (MDS) | 4 |
| Little Goose (LGS) and middle Snake (MDS) | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willis, S.C.; Parker, B.; Schreier, A.D.; Beamesderfer, R.; Miller, D.; Young, S.; Narum, S.R. Population Structure of White Sturgeon (Acipenser transmontanus) in the Columbia River Inferred from Single-Nucleotide Polymorphisms. Diversity 2022, 14, 1045. https://doi.org/10.3390/d14121045
Willis SC, Parker B, Schreier AD, Beamesderfer R, Miller D, Young S, Narum SR. Population Structure of White Sturgeon (Acipenser transmontanus) in the Columbia River Inferred from Single-Nucleotide Polymorphisms. Diversity. 2022; 14(12):1045. https://doi.org/10.3390/d14121045
Chicago/Turabian StyleWillis, Stuart C., Blaine Parker, Andrea D. Schreier, Ray Beamesderfer, Donella Miller, Shawn Young, and Shawn R. Narum. 2022. "Population Structure of White Sturgeon (Acipenser transmontanus) in the Columbia River Inferred from Single-Nucleotide Polymorphisms" Diversity 14, no. 12: 1045. https://doi.org/10.3390/d14121045