

Morphological and Molecular Characterization Using Genitalia and CoxI Barcode Sequence Analysis of Afrotropical Mosquitoes with Arbovirus Vector Potential
 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mosquito Sampling and Preparation of Male Mosquito Genitalia
2.2. DNA Extraction, Partial Amplification of CoxI, and Culex Pipiens Complex Molecular Identification
2.3. Amplicon Sequencing and Nucleotide Sequence Analyses
3. Results
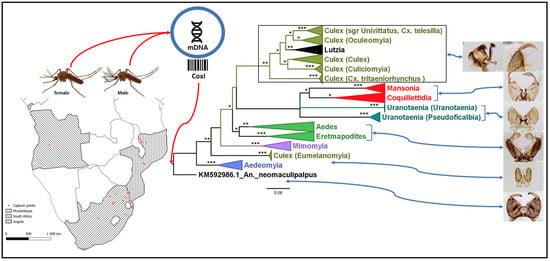
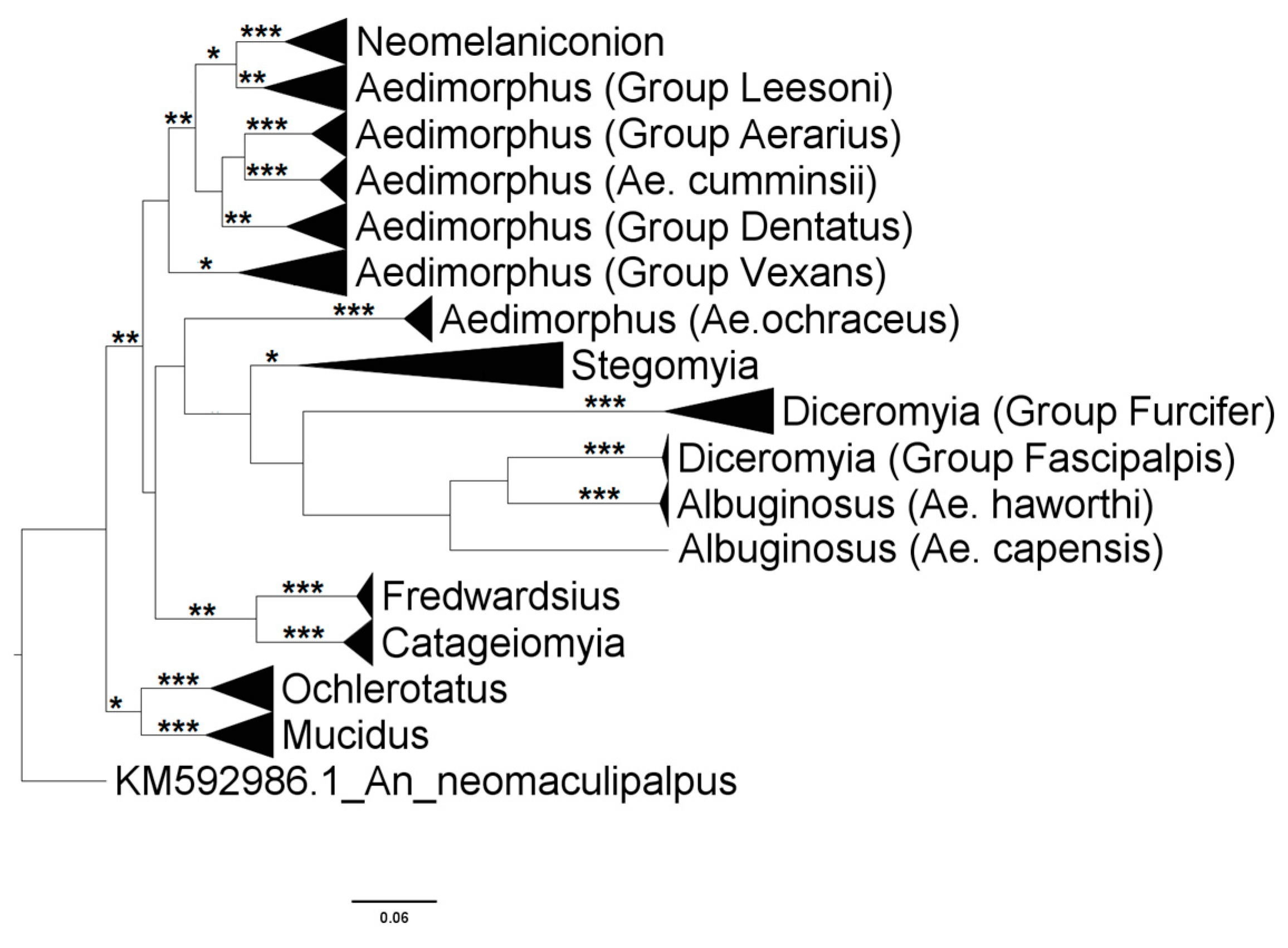
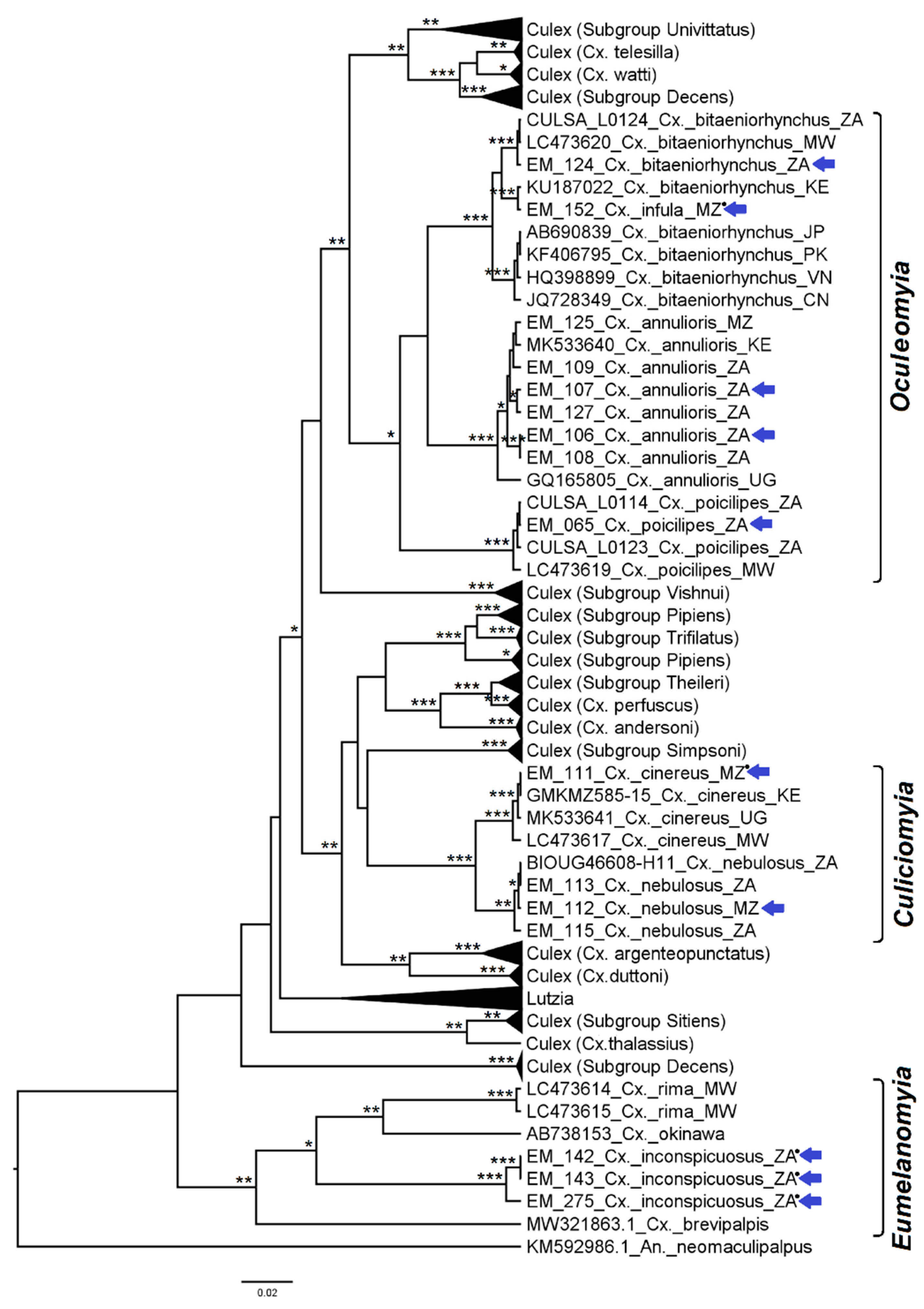
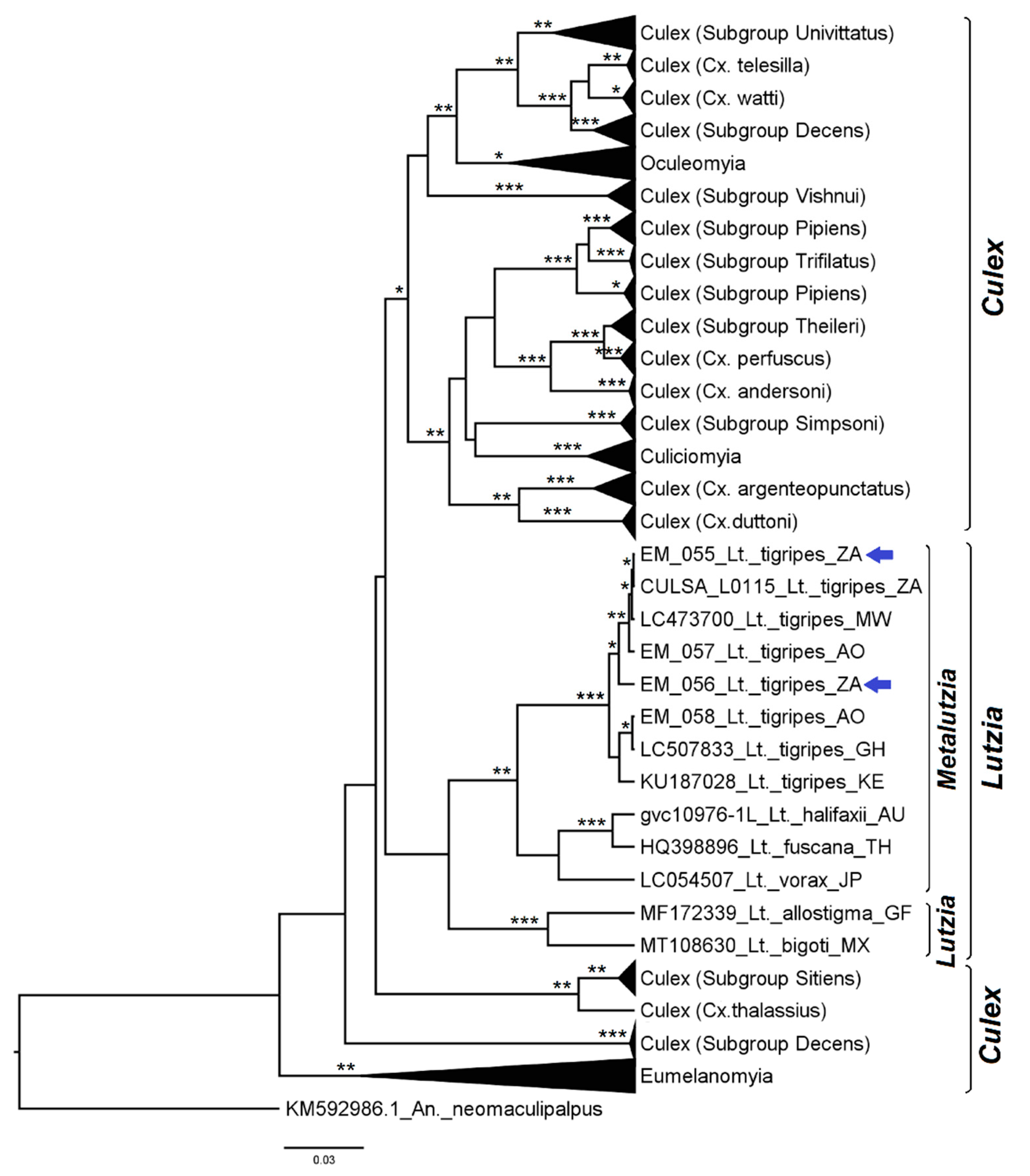
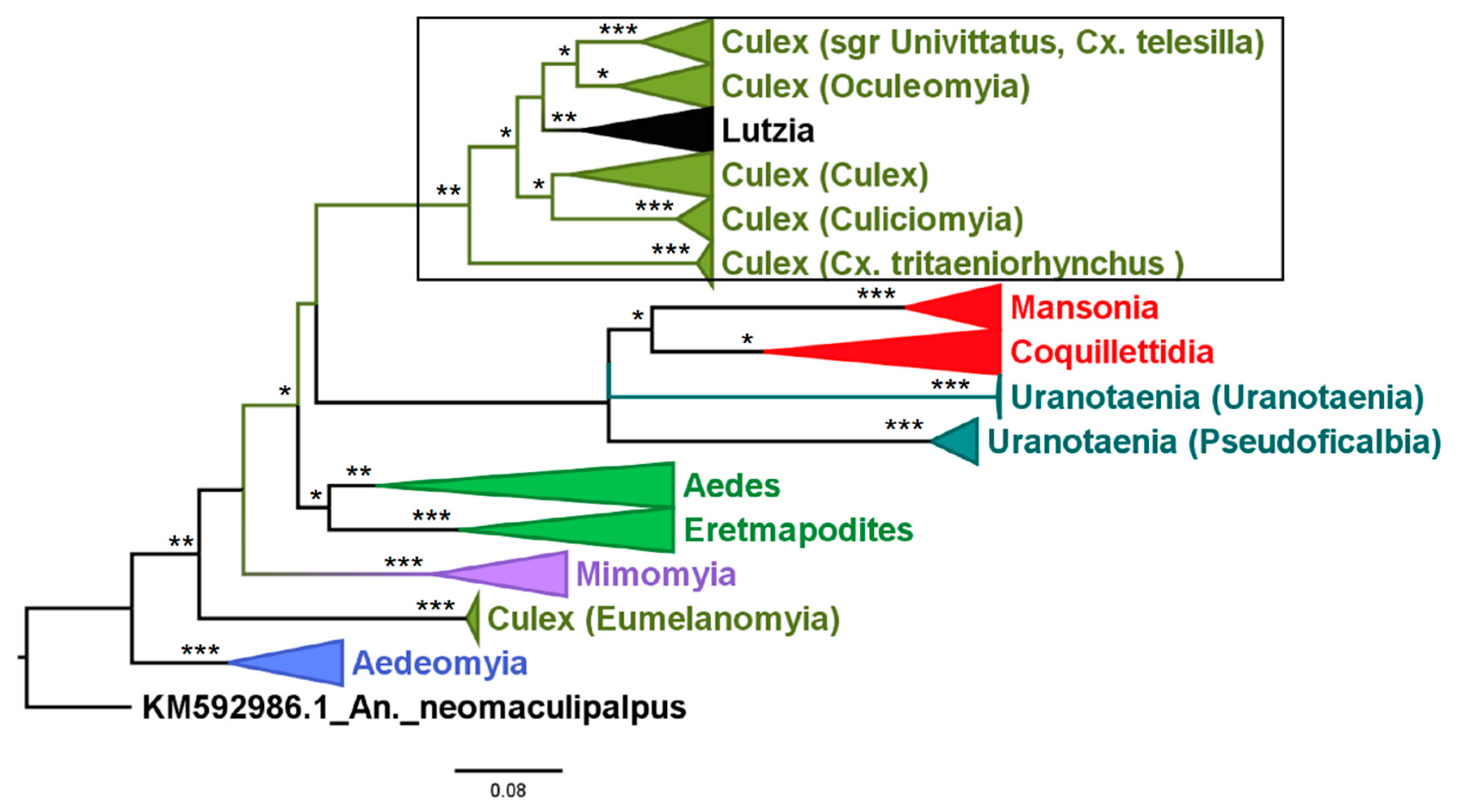
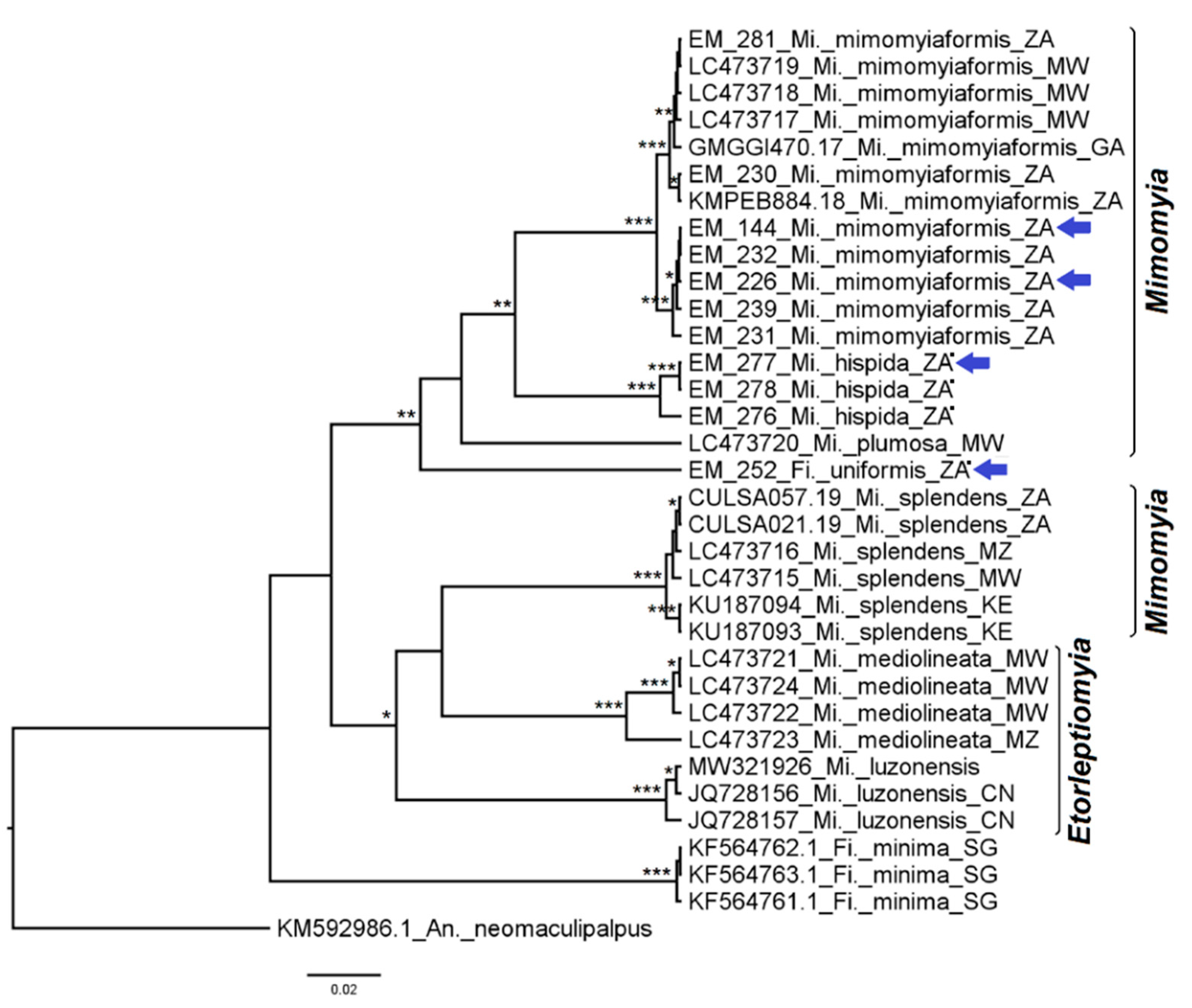
3.1. Mosquito Identification: Morphological and Molecular
3.2. Mosquito Identification Using Phylogenetic Reconstruction
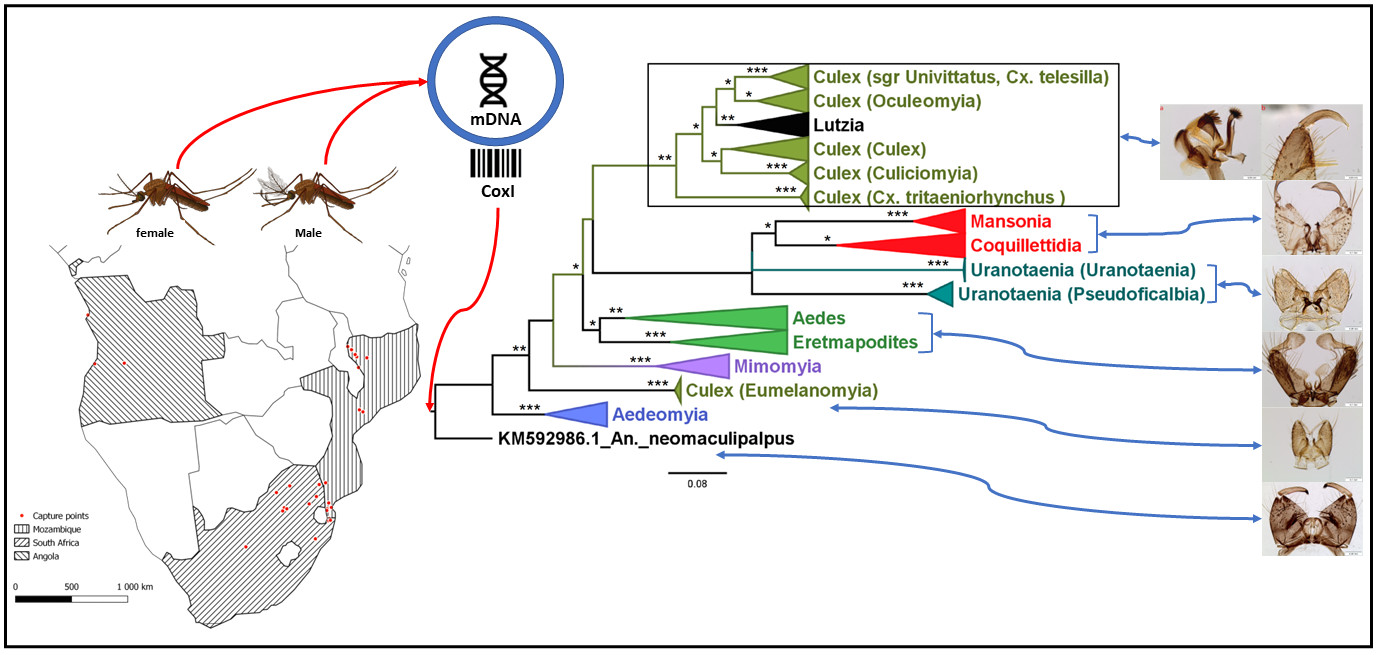
3.2.1. Genus Aedeomyia
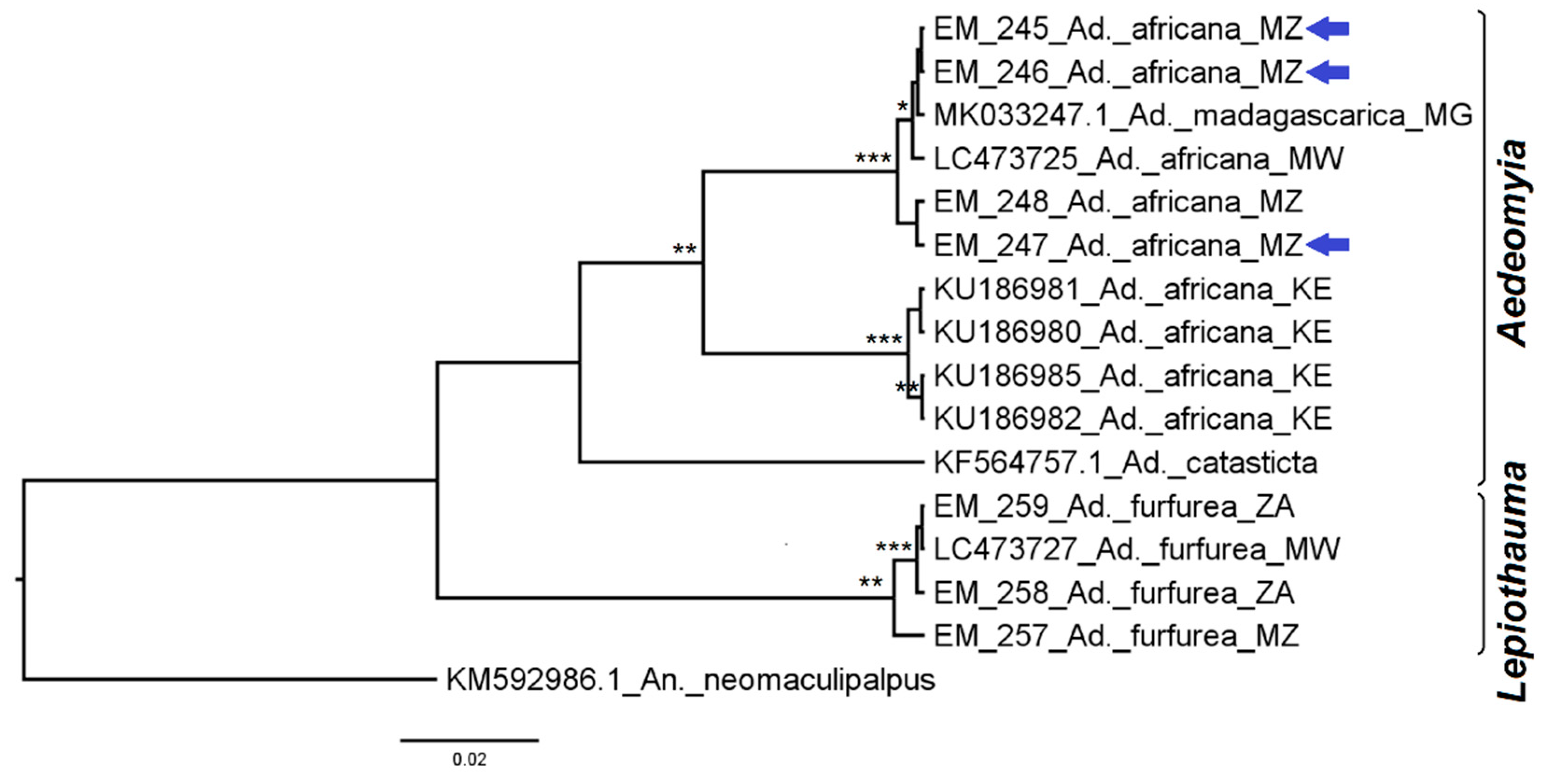
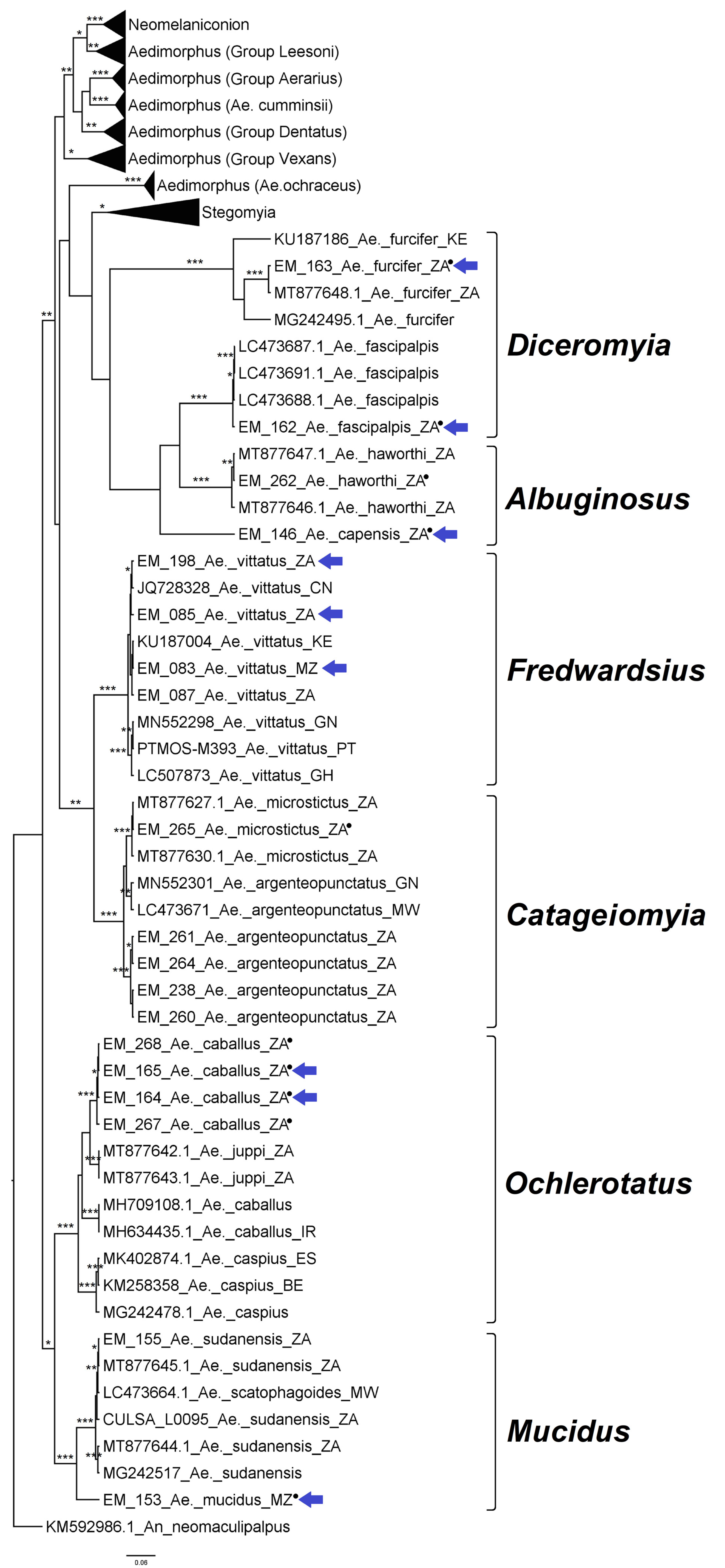
3.2.2. Genus Aedes
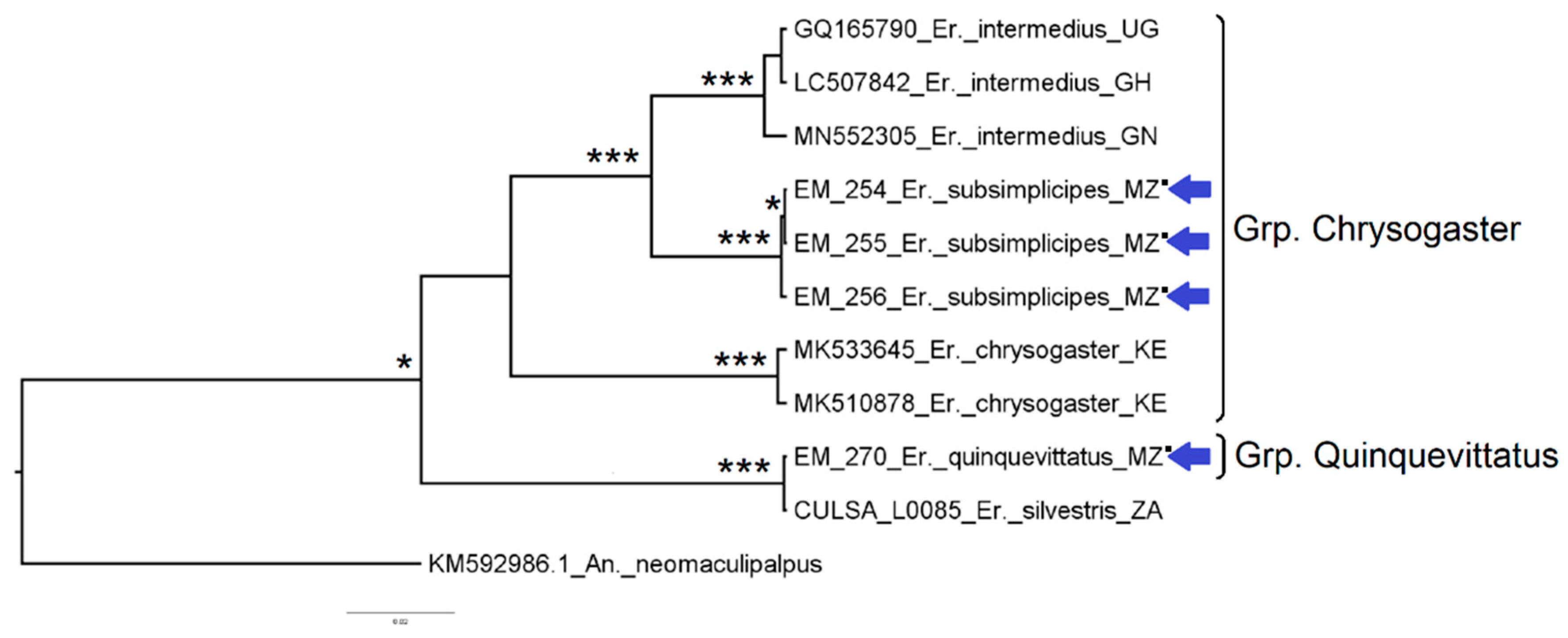
3.2.3. Genus Eretmapodites
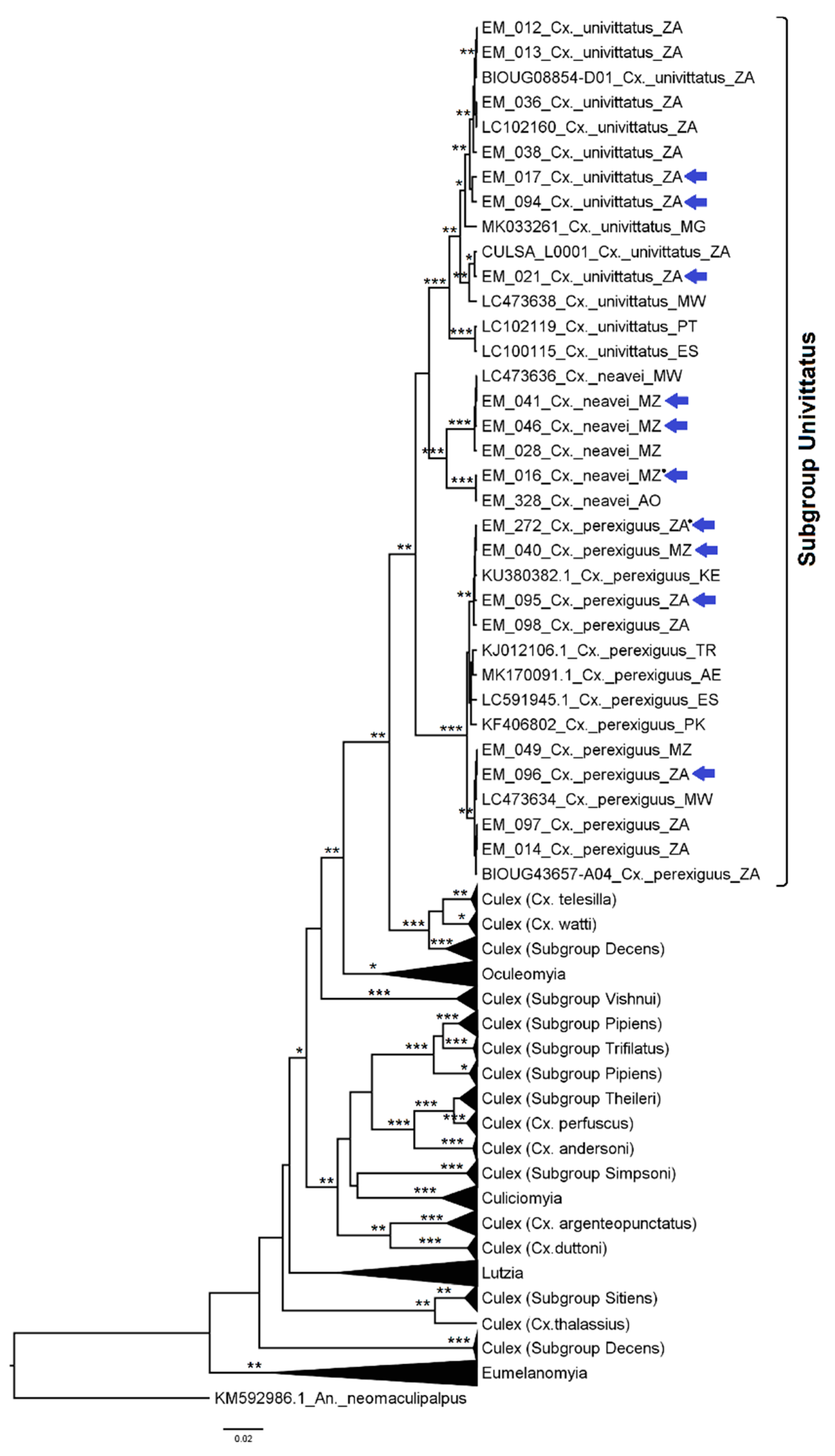
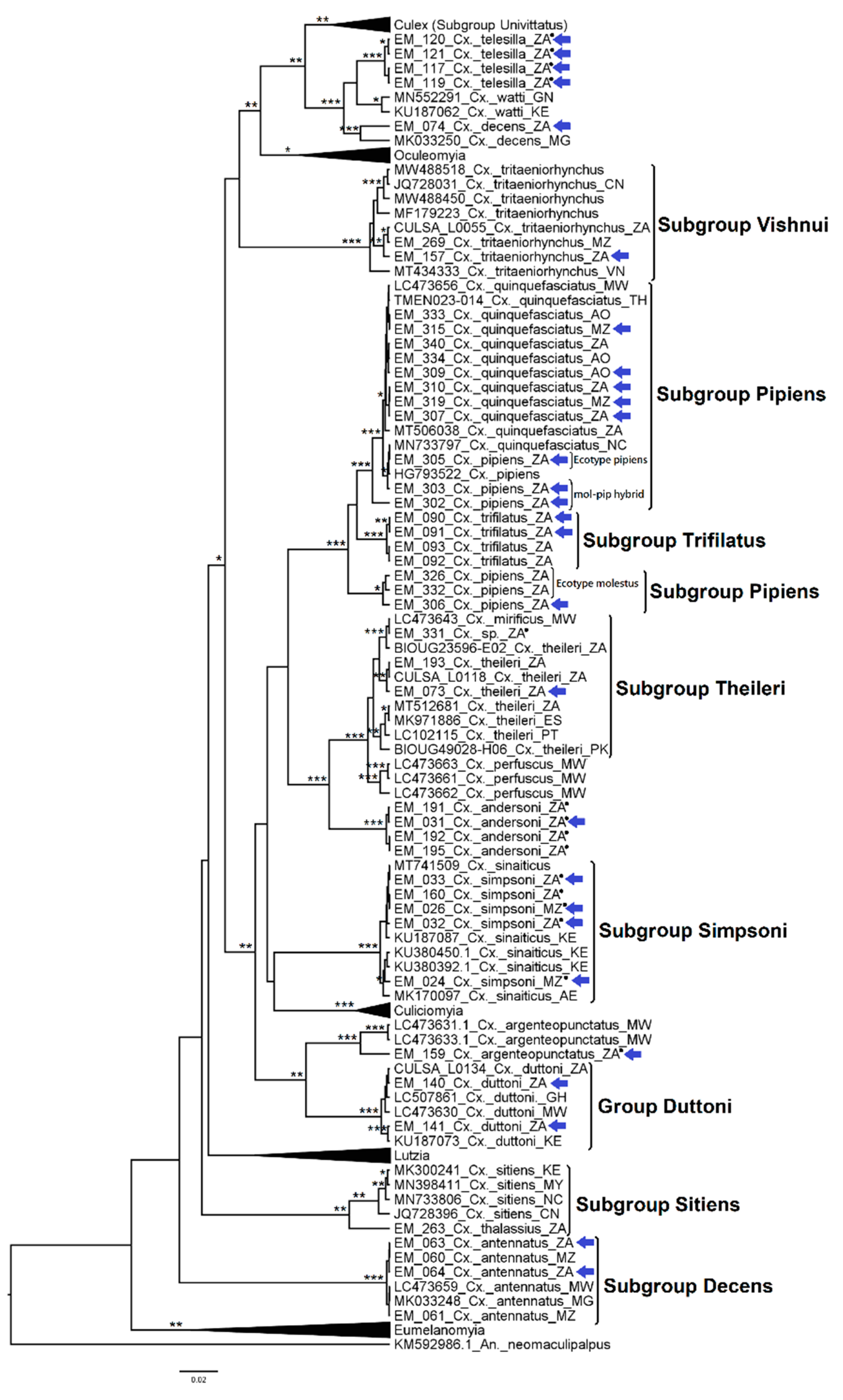
3.2.4. Genera Culex and Lutzia
3.2.5. Genera Ficalbia and Mimomyia
3.2.6. Genus Coquillettidia
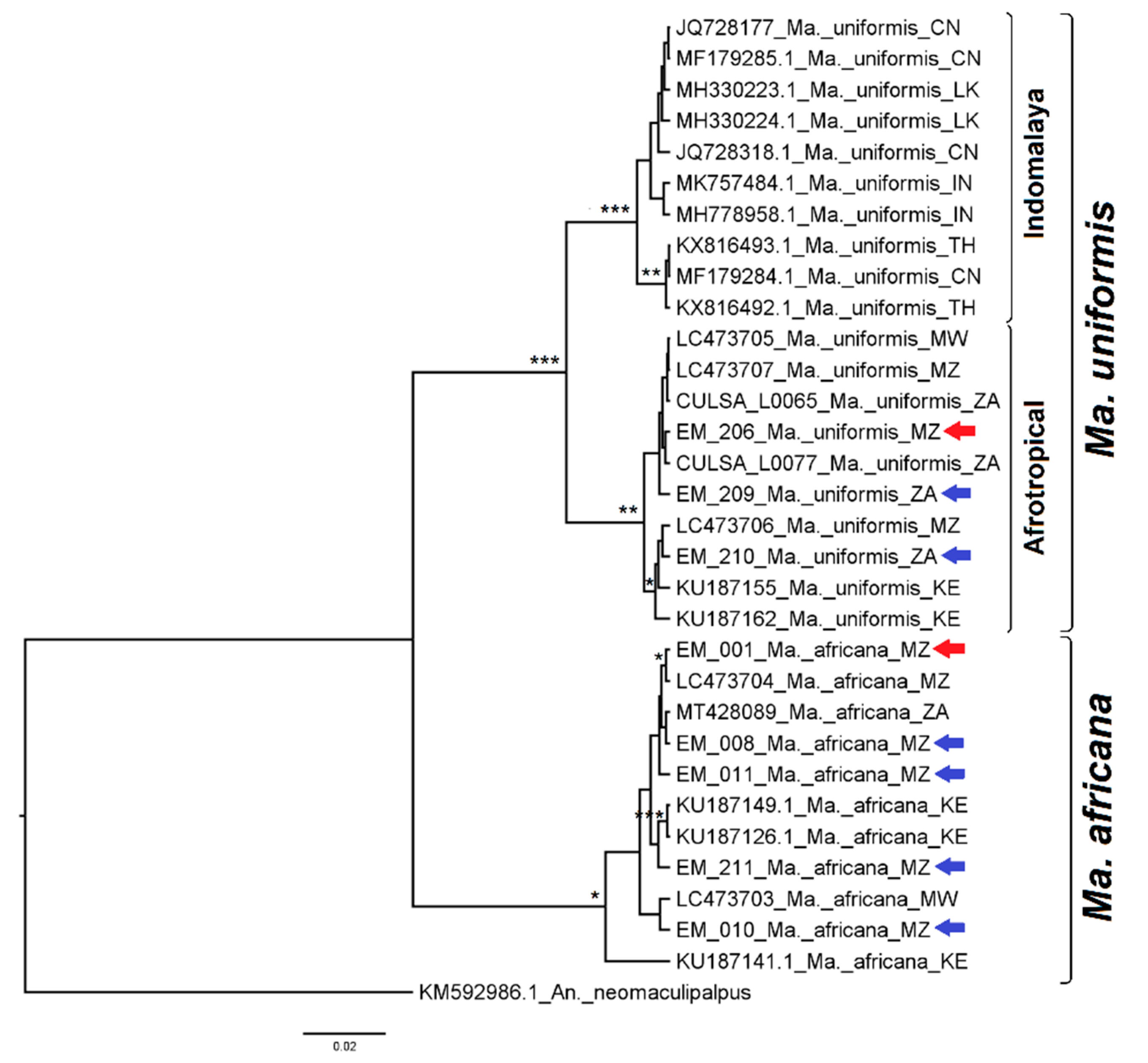
3.2.7. Genus Mansonia
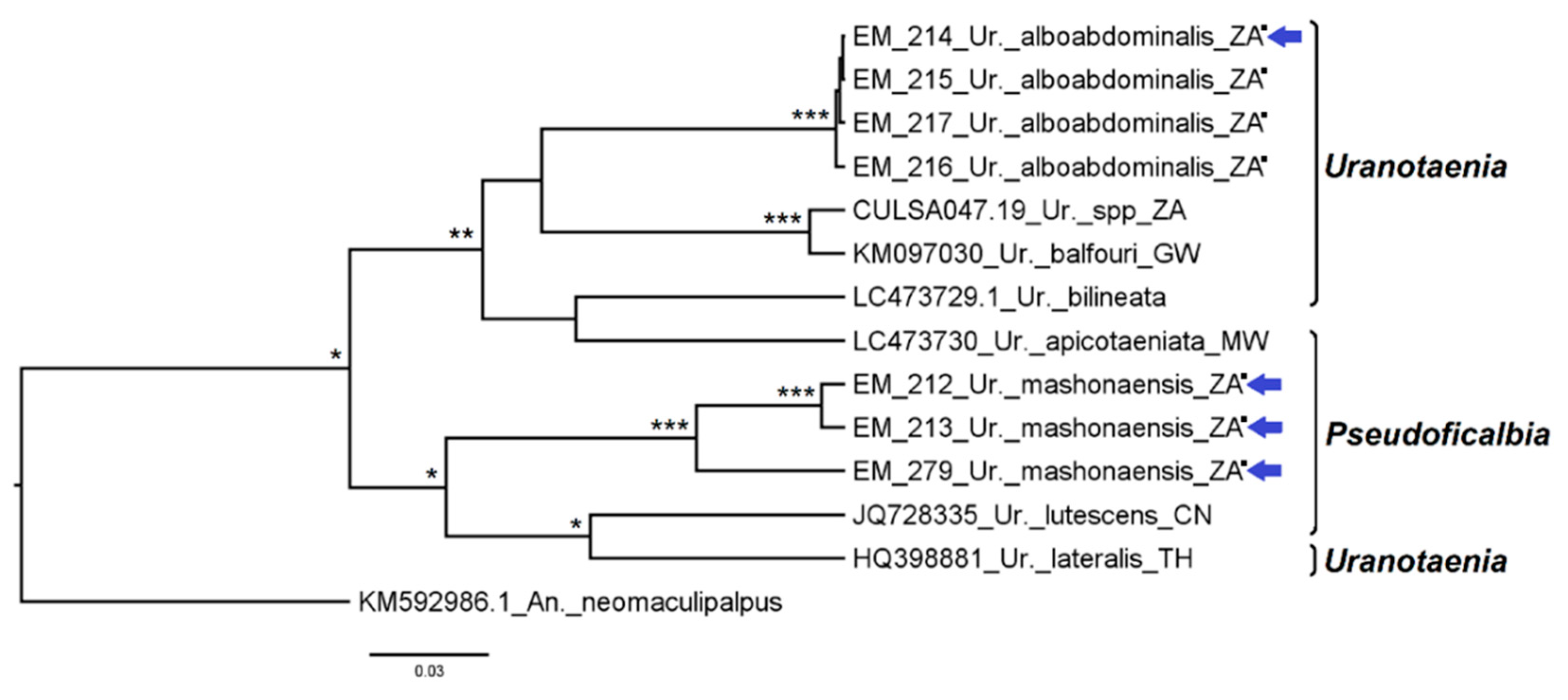
3.2.8. Genus Uranotaenia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilkerson, R.; Linton, Y.-M.; Strickman, D. Mosquitoes of the World; Johns Hopkins University Press: Baltimore, MA, USA, 2021; Volume 1. [Google Scholar]
- Braack, L.; De Almeida, A.P.G.; Cornel, A.J.; Swanepoel, R.; De Jager, C. Mosquito-Borne Arboviruses of African Origin: Review of Key Viruses and Vectors. Parasites Vectors 2018, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Venter, M. Assessing the Zoonotic Potential of Arboviruses of African Origin. Curr. Opin. Virol. 2018, 28, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, F.W. Mosquitoes of the Ethiopian Region. III.-Culicine Adults and PupAe; British Museum (N.H.): London, UK, 1941. [Google Scholar]
- Jupp, P.G. Mosquitoes of Southern Africa: Culicinae and Toxorhynchitinae; Ekogilde Publishers: New York, NY, USA, 1996. [Google Scholar]
- Huang, Y. Aedes (Stegomyia) Simpsoni Complex in the Ethiopian Region with Lectotype Designation for Simpsoni (Theobald) (Diptera: Culicidae). Mosq. Syst. 1979, 11, 221–234. [Google Scholar]
- Huang, Y. Aedes (Stegomyia) Bromeliae (Diptera: Culicidae), the Yellow Fever Virus Vector in East Africa. J. Med. Entomol. 1986, 23, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Notes on the Aedes (Diceromyia) Furcifer Group, with a Description of a New Species (Diptera: Culicidae). Proc. Entomol. Soc. Washingt. 1986, 88, 634–649. [Google Scholar]
- Harbach, R.E.; Knight, K.L. Taxonomists’ Glossary of Mosquito Anatomy; Plexus Publishing, Inc.: Marlton, NJ, USA, 1980; ISBN 9780937548004. [Google Scholar]
- Walton, C.; Sharpe, R.G.; Pritchard, S.J.; Thelwell, N.J.; Butlin, R.K. Molecular Identification of Mosquito Species. Biol. J. Linn. Soc. 1999, 68, 241–256. [Google Scholar] [CrossRef]
- Smith, J.L.; Fonseca, D.M. Rapid Assays for Identification of Members of the Culex (Culex) Pipiens Complex, Their Hybrids, and Other Sibling Species (Diptera: Culicidae). Am. J. Trop. Med. Hyg. 2004, 70, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Guarido, M.M.; Riddin, M.A.; Johnson, T.; Braack, L.E.O.; Schrama, M.; Gorsich, E.E.; Brooke, B.D.; Almeida, A.P.G.; Venter, M. Aedes Species (Diptera: Culicidae) Ecological and Host Feeding Patterns in the North-Eastern Parts of South Africa, 2014–2018. Parasites Vectors 2021, 14, 339. [Google Scholar] [CrossRef]
- Maekawa, Y.; Pemba, D.; Kumala, J.; Gowelo, S.; Higa, Y.; Futami, K.; Sawabe, K.; Tsuda, Y. DNA Barcoding of Mosquitoes Collected through a Nationwide Survey in 2011 and 2012 in Malawi, Southeast Africa. Acta Trop. 2021, 213, 105742. [Google Scholar] [CrossRef]
- Mixão, V.; Bravo Barriga, D.; Parreira, R.; Novo, M.T.; Sousa, C.A.; Frontera, E.; Venter, M.; Braack, L.; Almeida, A.P.G. Comparative Morphological and Molecular Analysis Confirms the Presence of the West Nile Virus Mosquito Vector, Culex Univittatus, in the Iberian Peninsula. Parasites Vectors 2016, 9, 601. [Google Scholar] [CrossRef] [Green Version]
- Makanda, M.; Kemunto, G.; Wamuyu, L.; Bargul, J.; Muema, J.; Mutunga, J. Diversity and Molecular Characterization of Mosquitoes (Diptera: Culicidae) in Selected Ecological Regions in Kenya. F1000Research 2018, 7, 262. [Google Scholar] [CrossRef] [Green Version]
- Ajamma, Y.U.; Villinger, J.; Omondi, D.; Salifu, D.; Onchuru, T.O.; Njoroge, L.; Muigai, A.W.T.; Masiga, D.K. Composition and Genetic Diversity of Mosquitoes (Diptera: Culicidae) on Islands and Mainland Shores of Kenya’s Lakes Victoria and Baringo. J. Med. Entomol. 2016, 53, 1348–1363. [Google Scholar] [CrossRef] [PubMed]
- Hemmerter, S.; Šlapeta, J.; Beebe, N.W. Resolving Genetic Diversity in Australasian Culex Mosquitoes: Incongruence between the Mitochondrial Cytochrome c Oxidase I and Nuclear Acetylcholine Esterase 2. Mol. Phylogenet. Evol. 2009, 50, 317–325. [Google Scholar] [CrossRef]
- Hernández-Triana, L.M.; Brugman, V.A.; Nikolova, N.I.; Ruiz-Arrondo, I.; Barrero, E.; Thorne, L.; de Marco, M.F.; Krüger, A.; Lumley, S.; Johnson, N.; et al. DNA Barcoding of British Mosquitoes (Diptera, Culicidae) to Support Species Identification, Discovery of Cryptic Genetic Diversity and Monitoring Invasive Species. Zookeys 2019, 2019, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Awedoba, A.K. West Africa. Afr. Yearb. 2020, 16, 32–47. [Google Scholar] [CrossRef]
- Chan, A.; Chiang, L.P.; Hapuarachchi, H.C.; Tan, C.H.; Pang, S.C.; Lee, R.; Lee, K.S.; Ng, L.C.; Lam-Phua, S.G. DNA Barcoding: Complementing Morphological Identification of Mosquito Species in Singapore. Parasites Vectors 2014, 7, 569. [Google Scholar] [CrossRef]
- Ashfaq, M.; Hebert, P.D.N.; Mirza, J.H.; Khan, A.M.; Zafar, Y.; Mirza, M.S. Analyzing Mosquito (Diptera: Culicidae) Diversity in Pakistan by DNA Barcoding. PLoS ONE 2014, 9, e97268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demari-Silva, B.; Vesgueiro, F.T.; Sallum, M.A.M.; Marrelli, M.T. Taxonomic and Phylogenetic Relationships between Species of the Genus Culex (Diptera: Culicidae) from Brazil Inferred from the Cytochrome c Oxidase I Mitochondrial Gene. J. Med. Entomol. 2011, 48, 272–279. [Google Scholar] [CrossRef]
- Kumar, N.P.; Rajavel, A.R.; Natarajan, R.; Jambulingam, P. DNA Barcodes Can Distinguish Species of Indian Mosquitoes (Diptera: Culicidae). J. Med. Entomol. 2007, 44, 1–7. [Google Scholar] [CrossRef]
- Laurito, M.; De Oliveira, T.M.P.; Almiron, W.R.; Sallum, M.A.M. COI Barcode versus Morphological Identification of Culex (Culex) (Diptera: Culicidae) Species: A Case Study Using Samples from Argentina and Brazil. Mem. Inst. Oswaldo Cruz 2013, 108, 110–122. [Google Scholar] [CrossRef]
- Pagac, B.B.; Spring, A.R.; Stawicki, J.R.; Dinh, T.L.; Lura, T.; Kavanaugh, M.D.; Pecor, D.B.; Justi, S.A.; Linton, Y.M. Incursion and Establishment of the Old World Arbovirus Vector Aedes (Fredwardsius) Vittatus (Bigot, 1861) in the Americas. Acta Trop. 2021, 213, 105739. [Google Scholar] [CrossRef] [PubMed]
- Talaga, S.; Leroy, C.; Guidez, A.; Dusfour, I.; Girod, R.; Dejean, A.; Murienne, J. DNA Reference Libraries of French Guianese Mosquitoes for Barcoding and Metabarcoding. PLoS ONE 2017, 12, e0176993. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, C.; Guo, X.; Xing, D.; Dong, Y.; Wang, Z.; Zhang, Y.; Liu, M.; Zheng, Z.; Zhang, H.; et al. Identifying the Main Mosquito Species in China Based on DNA Barcoding. PLoS ONE 2012, 7, e47051. [Google Scholar] [CrossRef] [PubMed]
- Weeraratne, T.C.; Surendran, S.N.; Karunaratne, S.H.P.P. DNA Barcoding of Morphologically Characterized Mosquitoes Belonging to the Subfamily Culicinae from Sri Lanka. Parasites Vectors 2018, 11, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.; Braack, L.; Guarido, M.; Venter, M.; Gouveia Almeida, A.P. Mosquito Community Composition and Abundance at Contrasting Sites in Northern South Africa, 2014–2017. J. Vector Ecol. 2020, 45, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Abílio, A.P.; Silva, M.; Kampango, A.; Narciso, I.; Gudo, E.S.; Das Neves, L.C.B.; Sidat, M.; Fafetine, J.M.; De Almeida, A.P.G.; Parreira, R. A Survey of RNA Viruses in Mosquitoes from Mozambique Reveals Novel Genetic Lineages of Flaviviruses and Phenuiviruses, as Well as Frequent Flavivirus-like Viral DNA Forms in Mansonia. BMC Microbiol. 2020, 20, 225. [Google Scholar] [CrossRef]
- Guarido, M.M.; Motlou, T.; Riddin, M.A.; MacIntyre, C.; Manyana, S.C.; Johnson, T.; Schrama, M.; Gorsich, E.E.; Brooke, B.D.; Almeida, A.G.P.; et al. Potential Mosquito Vectors for Shuni Virus, South Africa, 2014–2018. Emerg. Infect. Dis. 2021, 27, 3142–3146. [Google Scholar] [CrossRef]
- Guarido, M.M.; Govender, K.; Riddin, M.A.; Schrama, M.; Gorsich, E.E.; Brooke, B.D.; Almeida, A.P.G.; Venter, M. Detection of Insect-Specific Flaviviruses in Mosquitoes (Diptera: Culicidae) in Northeastern Regions of South Africa. Viruses 2021, 13, 2148. [Google Scholar] [CrossRef]
- Marrelli, M.T.; Sallum, M.A.M.; Marinotti, O. The Second Internal Transcribed Spacer of Nuclear Ribosomal DNA as a Tool for Latin American Anopheline Taxonomy—A Critical Review. Mem. Inst. Oswaldo Cruz 2006, 101, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Harbach, R.E. The Mosquitoes of the Subgenus Culex in Southwestern Asia and Egypt (Diptera: Culicidae). Contrib. Am. Entomol. Inst. 1988, 24, 1. [Google Scholar]
- Wilkerson, R.C.; Linton, Y.M.; Fonseca, D.M.; Schultz, T.R.; Price, D.C.; Strickman, D.A. Making Mosquito Taxonomy Useful: A Stable Classification of Tribe Aedini That Balances Utility with Current Knowledge of Evolutionary Relationships. PLoS ONE 2015, 10, e0133602. [Google Scholar] [CrossRef] [PubMed]
- Sirivanakarn, S. Medical Entomology Studies—A Revision of the Subgenus Culex in the Oriental Region (Diptera: Culicidae). Contrib. Am. Entomol. Inst. 1976, 12, 1–272. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA Primers for Amplification of Mitochondrial Cytochrome c Oxidase Subunit I from Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Tchouassi, D.P.; Bastos, A.D.S.; Sole, C.L.; Diallo, M.; Lutomiah, J.; Mutisya, J.; Mulwa, F.; Borgemeister, C.; Sang, R.; Torto, B. Population Genetics of Two Key Mosquito Vectors of Rift Valley Fever Virus Reveals New Insights into the Changing Disease Outbreak Patterns in Kenya. PLoS Negl. Trop. Dis. 2014, 8, e3364. [Google Scholar] [CrossRef]
- Bahnck, C.M.; Fonseca, D.M. Rapid Assay to Identify the Two Genetic Forms of Culex (Culex) Pipiens L. (Diptera: Culicidae) and Hybrid Populations. Am. J. Trop. Med. Hyg. 2006, 75, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; von Haeseler, A. Maximum-Likelihood Analysis Using TREE-PUZZLE. Curr. Protoc. Bioinform. 2007, 17, 6.6.1–6.6.23. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Bryant, D.; Moulton, V. Neighbor-Net: An Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Dress, A.W.M.; Huson, D.H. Constructing Splits Graphs. IEEE/ACM Trans. Comput. Biol. Bioinf. 2004, 1, 109–115. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, B.M. A Taxonomic Revision of Certain Aedes Species (Diptera: Culicidae) of the Subgenus Aedimorphus in Southern Africa. J. Entomol. Soc. S. Afr. 1975, 38, 251–287. [Google Scholar]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Torres-Gutierrez, C.; Bergo, E.S.; Emerson, K.J.; de Oliveira, T.M.P.; Greni, S.; Sallum, M.A.M. Mitochondrial COI Gene as a Tool in the Taxonomy of Mosquitoes Culex Subgenus Melanoconion. Acta Trop. 2016, 164, 137–149. [Google Scholar] [CrossRef]
- Diagne, C.T.; Diallo, D.; Faye, O.; Ba, Y.; Faye, O.; Gaye, A.; Dia, I.; Faye, O.; Weaver, S.C.; Sall, A.A.; et al. Potential of Selected Senegalese Aedes Spp. Mosquitoes (Diptera: Culicidae) to Transmit Zika Virus. BMC Infect. Dis. 2015, 15, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Harbach, R.E.; Howard, T.M. Corrections in the Status and Rank of Names Used to Denote Varietal Forms of Mosquitoes (Diptera: Culicidae). Zootaxa 2007, 48, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA Barcoding and Taxonomy in Diptera: A Tale of High Intraspecific Variability and Low Identification Success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kengne, P.; Goff, G.L.; Fontenille, D. Molecular Genetic Investigation of Morphological Species of Members of the Neomelaniconion Subgenus (Diptera: Culicidae: Aedini) from Madagascar Using Ribosomal Internal Transcribed Spacer 2. J. Med. Entomol. 2009, 46, 403–407. [Google Scholar] [CrossRef]
- Cornel, A.J.; Mcabee, R.D.; Rasgon, J.; Stanich, M.A.; Scott, T.W.; Coetzee, M. Differences in Extent of Genetic Introgression between Sympatric Culex Pipiens and Culex Quinquefasciatus (Diptera: Culicidae) in California and South Africa. J. Med. Entomol. 2003, 40, 36–51. [Google Scholar] [CrossRef]
- Gomes, B.; Alves, J.; Sousa, C.A.; Santa-Ana, M.; Vieira, I.; Silva, T.L.; Almeida, A.P.G.; Donnelly, M.J.; Pinto, J. Hybridization and Population Structure of the Culex Pipiens Complex in the Islands of Macaronesia. Ecol. Evol. 2012, 2, 1889–1902. [Google Scholar] [CrossRef]
- Gomes, B.; Sousa, C.A.; Novo, M.T.; Freitas, F.B.; Alves, R.; Côrte-Real, A.R.; Salgueiro, P.; Donnelly, M.J.; De Almeida, A.P.G.; Pinto, J. Asymmetric Introgression between Sympatric Molestus and Pipiens Forms of Culex Pipiens (Diptera: Culicidae) in the Comporta Region, Portugal. BMC Evol. Biol. 2009, 9, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Molaei, G.; Andreadis, T.G. Genetic Insights into the Population Structure of Culex Pipiens (Diptera: Culicidae) in the Northeastern United States by Using Microsatellite Analysis. Am. J. Trop. Med. Hyg. 2008, 79, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Amraoui, F.; Tijane, M.; Sarih, M.; Failloux, A.B. Molecular Evidence of Culex Pipiens Form Molestus and Hybrids Pipiens/Molestus in Morocco, North Africa. Parasites Vectors 2012, 5, 2010–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikevich, E.V.; Vinogradova, E.B.; Bouattour, A.; De Almeida, A.P.G. Genetic Diversity of Culex Pipiens Mosquitoes in Distinct Populations from Europe: Contribution of Cx. Quinquefasciatus in Mediterranean Populations. Parasites Vectors 2016, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, H. Research on the Mosquitoes of Angola (Diptera: Culicidae): II–Some New Culicine Records. An. Do Inst. Med. Trop. 1966, 23, 163–166. [Google Scholar]
- Weetman, D.; Kamgang, B.; Badolo, A.; Moyes, C.L.; Shearer, F.M.; Coulibaly, M.; Pinto, J.; Lambrechts, L.; McCall, P.J. Aedes Mosquitoes and Aedes-Borne Arboviruses in Africa: Current and Future Threats. Int. J. Environ. Res. Public Health 2018, 15, 220. [Google Scholar] [CrossRef]
- Moore, M.; Sylla, M.; Goss, L.; Burugu, M.W.; Sang, R.; Kamau, L.W.; Kenya, E.U.; Bosio, C.; de Munoz, M.L.; Sharakova, M.; et al. Dual African Origins of Global Aedes Aegypti s.l. Populations Revealed by Mitochondrial DNA. PLoS Negl. Trop. Dis. 2013, 7, e2175. [Google Scholar] [CrossRef] [Green Version]
- Gloria-Soria, A.; Ayala, D.; Bheecarry, A.; Calderon-Arguedas, O.; Chadee, D.D.; Chiappero, M.; Coetzee, M.; Elahee, K.B.; Fernandez-Salas, I.; Kamal, H.A.; et al. Global Genetic Diversity of Aedes Aegypti. Mol. Ecol. 2016, 25, 5377–5395. [Google Scholar] [CrossRef] [Green Version]
- da Silva, A.F.; Machado, L.C.; de Paula, M.B.; da Silva, P.V.C.J.; de Morais, B.R.V.; de Melo, S.M.A.V.; Wallau, G.L. Culicidae Evolutionary History Focusing on the Culicinae Subfamily Based on Mitochondrial Phylogenomics. Sci. Rep. 2020, 10, 18823. [Google Scholar] [CrossRef]
- Harbach, R.E. Classification within the Cosmopolitan Genus Culex (Diptera: Culicidae): The Foundation for Molecular Systematics and Phylogenetic Research. Acta Trop. 2011, 120, 1–14. [Google Scholar] [CrossRef]
- Cornel, A.J.; Mayi, M.P.A.; Kowo, C.; Foncha, D.; Andongma, E.; Anong, D.N.; Elad, M.; Djomo, C.; Tchuinkam, T.; Brisco, K.K.; et al. New Species of Culex (Culiciomyia) (Diptera: Culicidae) from Talangaye Forest in Cameroon and Descriptions and Identification Keys for Males of the Afrotropical Species of the Subgenus. Zootaxa 2020, 4858, zootaxa-4858. [Google Scholar] [CrossRef]
- Vesgueiro, F.T.; Demari-Silva, B.; dos Santos, M.R.; Sallum, M.A.M.; Marrelli, M.T. Intragenomic Variation in the Second Internal Transcribed Spacer of the Ribosomal DNA of Species of the Genera Culex and Lutzia (Diptera: Culicidae). Mem. Inst. Oswaldo Cruz 2011, 106, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitching, I.J.; Culverwell, C.L.; Harbach, R.E. The Phylogenetic Conundrum of Lutzia (Diptera: Culicidae: Culicini): A Cautionary Account of Conflict and Support. Insect Syst. Evol. 2015, 46, 269–290. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, T.J.; Fu, W.B.; Yan, Z.T.; Si, F.L.; Zhang, Y.J.; Mao, Q.M.; Demari-Silva, B.; Chen, B. The Complete Mt Genomes of Lutzia Halifaxia, Lt. Fuscanus and Culex Pallidothorax (Diptera: Culicidae) and Comparative Analysis of 16 Culex and Lutzia Mt Genome Sequences. Parasites Vectors 2019, 12, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbach, R.E.; Kitching, I.J.; Culverwell, C.L.; Dubois, J.; Linton, Y.M. Phylogeny of Mosquitoes of Tribe Culicini (Diptera: Culicidae) Based on Morphological Diversity. Zool. Scr. 2012, 41, 499–514. [Google Scholar] [CrossRef]
- van den Bergh, C.; Thompson, P.N.; Swanepoel, R.; Almeida, A.P.G.; Paweska, J.T.; van Vuren, P.J.; Wilson, W.C.; Kemp, A.; Venter, E.H. Detection of Rift Valley Fever Virus in Aedes (Aedimorphus) Durbanensis, South Africa. Pathogens 2022, 11, 125. [Google Scholar] [CrossRef]
- Andreeva, Y.V.; Khrabrova, N.V.; Alekseeva, S.S.; Abylkassymova, G.M.; Simakova, A.V.; Sibataev, A.K. First Record of the Invasive Mosquito Species Aedes Koreicus (Diptera, Culicidae) in the Republic of Kazakhstan. Parasite 2021, 28, 52. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalvo-Sabino, E.; Abílio, A.P.; Guarido, M.M.; Valadas, V.; Novo, M.T.; Kampango, A.; Sousa, C.A.; Fafetine, J.; Venter, M.; Thompson, P.N.; et al. Morphological and Molecular Characterization Using Genitalia and CoxI Barcode Sequence Analysis of Afrotropical Mosquitoes with Arbovirus Vector Potential. Diversity 2022, 14, 940. https://doi.org/10.3390/d14110940
Montalvo-Sabino E, Abílio AP, Guarido MM, Valadas V, Novo MT, Kampango A, Sousa CA, Fafetine J, Venter M, Thompson PN, et al. Morphological and Molecular Characterization Using Genitalia and CoxI Barcode Sequence Analysis of Afrotropical Mosquitoes with Arbovirus Vector Potential. Diversity. 2022; 14(11):940. https://doi.org/10.3390/d14110940
Chicago/Turabian StyleMontalvo-Sabino, Eddyson, Ana Paula Abílio, Milehna Mara Guarido, Vera Valadas, Maria Teresa Novo, Ayubo Kampango, Carla Alexandra Sousa, José Fafetine, Marietjie Venter, Peter N. Thompson, and et al. 2022. "Morphological and Molecular Characterization Using Genitalia and CoxI Barcode Sequence Analysis of Afrotropical Mosquitoes with Arbovirus Vector Potential" Diversity 14, no. 11: 940. https://doi.org/10.3390/d14110940