
Introduced, Mixed, and Peripheral: Conservation of Mitochondrial-DNA Lineages in the Wild Boar (Sus scrofa L.) Population in the Urals

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Data Analysis
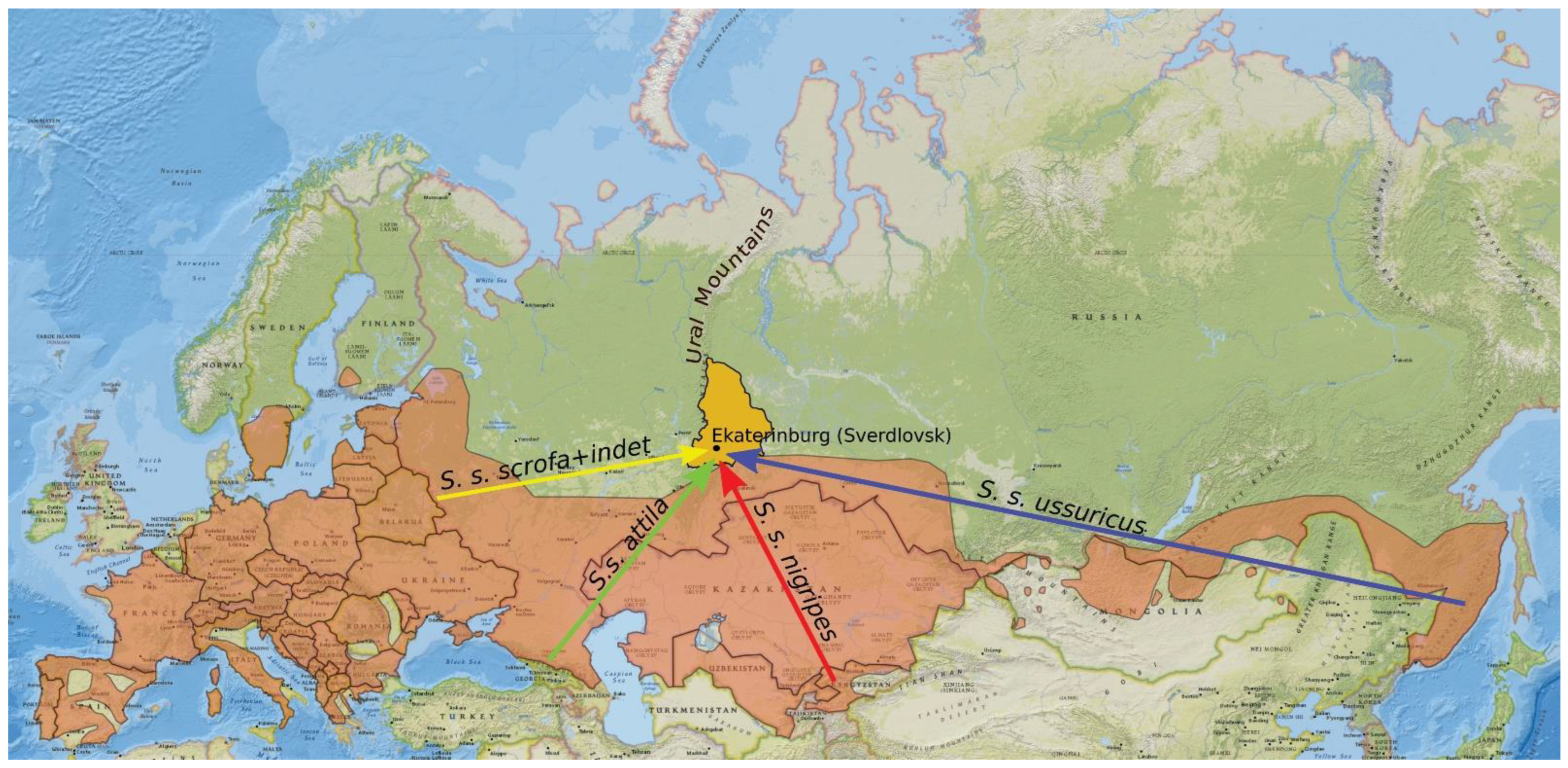
2.4. Collection of Data on Animals Released in the Urals
3. Results
3.1. Genetic Diversity and Haplotype Composition of the Introduced versus Founder Populations
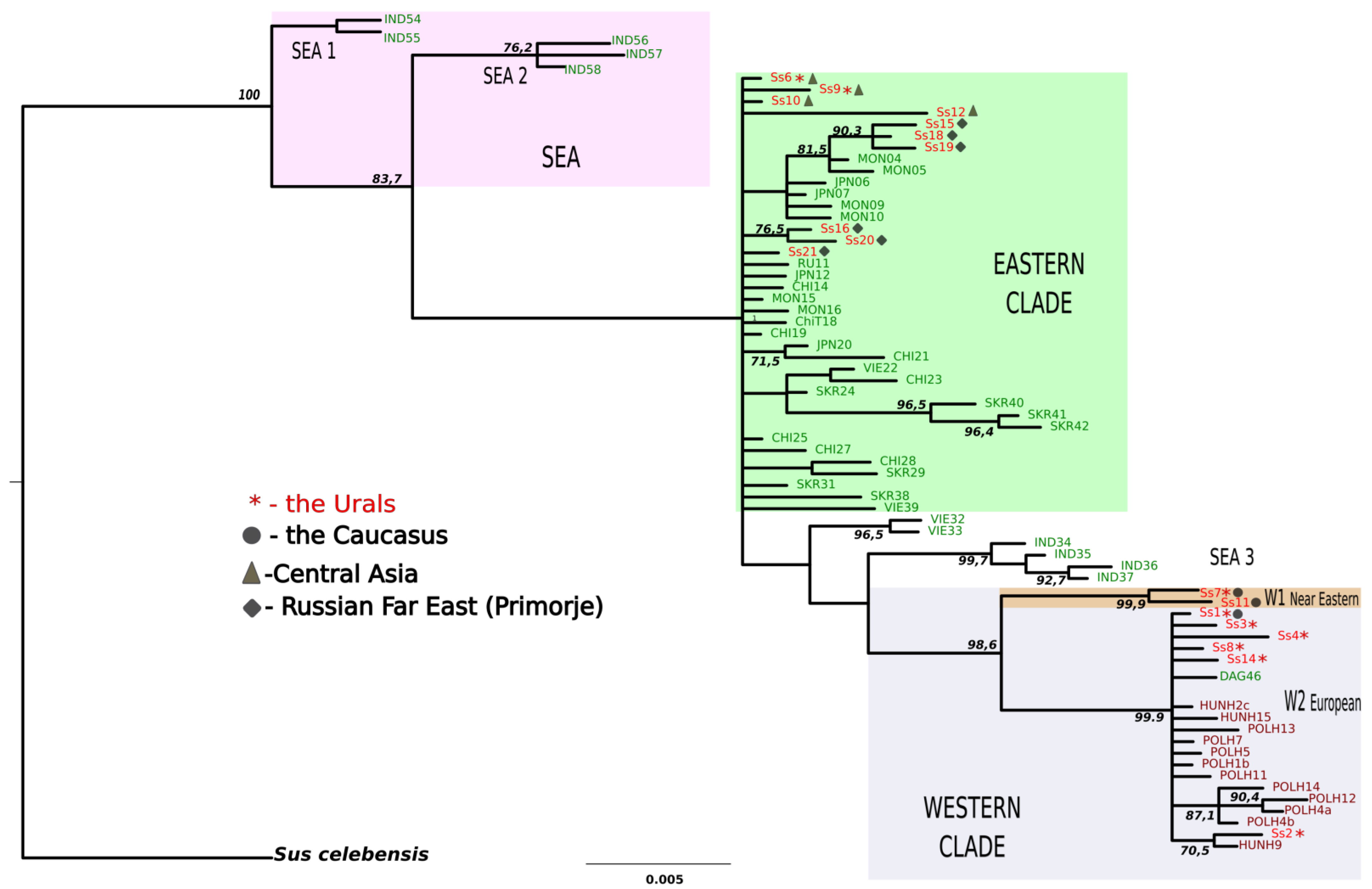
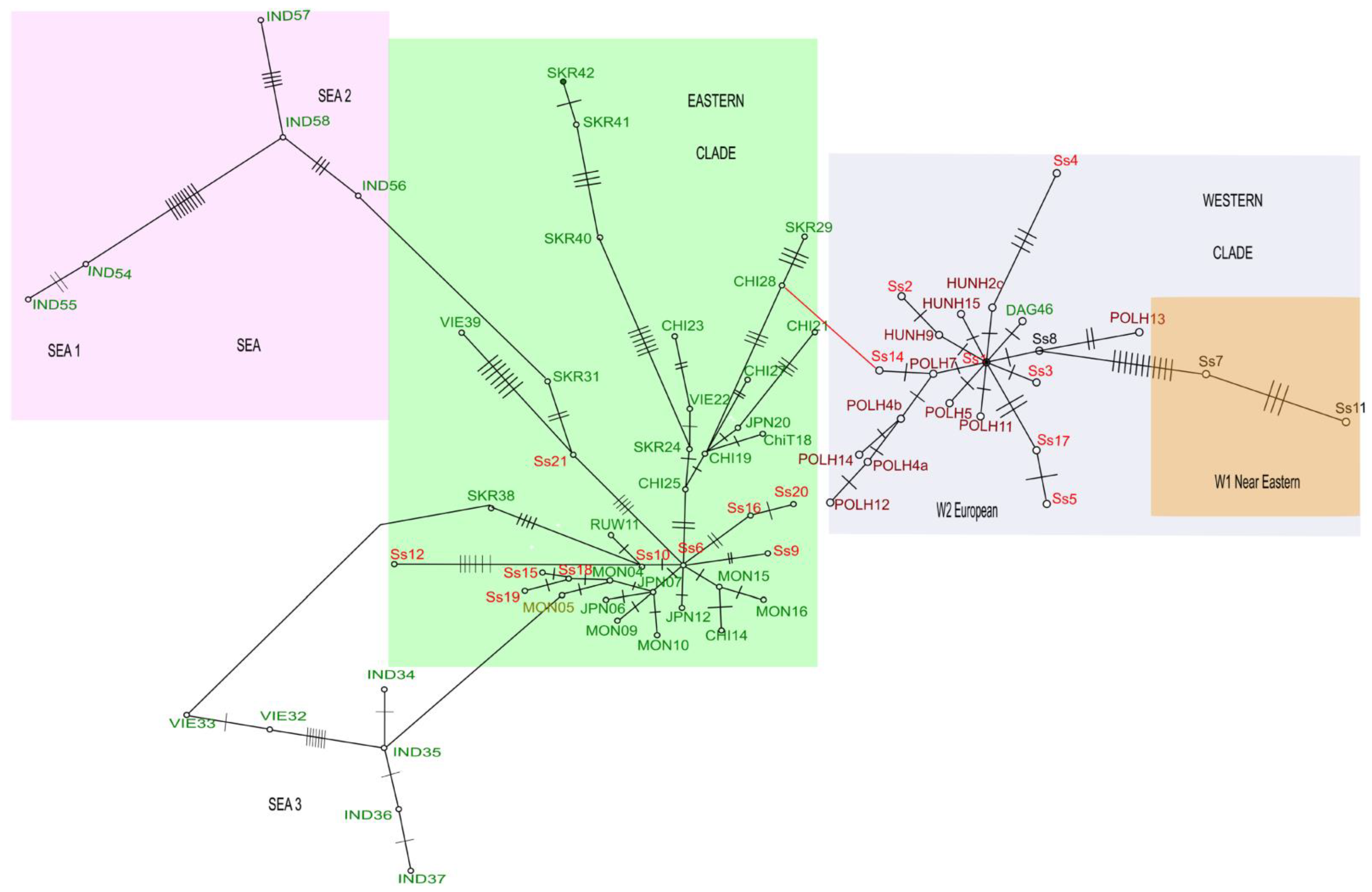
3.2. Phylogenetic Status of the Introduced versus Founder Populations
3.2.1. The Topology of Trees
3.2.2. Positions of Founder Populations on the Phylogenetic Tree
3.2.3. Positions of Haplotypes from the Introduced Population on the Phylogenetic Tree
3.3. Proportions of MtDNA Lineages in the Current Wild Boar Population of the Urals in Comparison with the Proportions of Released Subspecies
4. Discussion
4.1. Genetic Diversity in the Introduced Population
4.2. Phylogenetic Lineages in the Natural and Introduced Populations
4.3. Predominance of the Western Clade and a Decline in Eastern Mitochondrial Lineages in the Introduced Population of Wild Boars
- Animals from Asia were not detected in the sampled region because they dispersed to some other territories. Validation of this explanation requires wider sampling across the whole Ural region.
- The proportion of European haplotypes increased due to unofficial releases in the 2000s. Neither the number of individuals released during such unofficial introductions nor their origin are known. It is unlikely that wild boars were brought to the Urals from distant regions of Russia (such as the Russian Far East or Caucasus) or from Central Asia. Most probably, they were taken from local populations or brought from mixed populations of Central Russia, where the European lineage is more prevalent than the Asian one [69]. They could also be hybrids between wild boars and domestic pigs. All these factors could affect the genetic composition of the introduced population in some way. On the other hand, the current population of wild boars was established well before the 2000s. Thus, we believe that the haplotype frequencies in the current population are mainly a consequence of natural processes.
- Our third explanation is that animals carrying Western mitochondrial haplotypes are better adapted for survival and expansion in a new environment than those from the Eastern haplogroup. The lower survival rate of Eastern (particularly Far Eastern) haplotypes could be caused by two processes. First, the animals from the Caucasus, Central Asia, and Russian Far East could disperse across long distances from the release site. A possible reason is a difference in the habitat composition between the Urals and the regions of the animals’ origin (natal habitat preference induction hypothesis [105]). More distant dispersal could lead to higher mortality [106] and hence to a decrease in the proportion of Asian haplotypes. The second process is related to the genetic diversity in the founder populations. Recently, Kostyunina O. et al. [70] showed lower genomic variation in Far Eastern wild boars than in European ones. It is possible that the low genetic variation could cause low adaptability of Far Eastern wild boars, which, in turn, leads to the elimination of their mitochondrial haplotypes. Moreover, Tsai T.S. et al. [107] demonstrated that domestic pigs with European mitochondrial haplotypes produce significantly larger litters, whereas the haplotypes that cluster with Asian mitochondrial haplotypes have the lowest reproductive efficiency rates. The higher proportion and adaptability of the European lineage could also be attributed to a relatively high level of hybridization with domestic pigs in many European populations [108].
4.4. The Wild Boar in the Urals as a Model of Genetic Processes in Expanding (or Invading) Populations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ehrenfeld, J.G. Ecosystem Consequences of Biological Invasions. Annu. Rev. Ecol. Evol. Syst. 2010, 41, 59–80. [Google Scholar] [CrossRef] [Green Version]
- Dlugosch, K.M.; Parker, I.M. Founding Events in Species Invasions: Genetic Variation, Adaptive Evolution, and the Role of Multiple Introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.; Darling, J. Paradox Lost: Genetic Diversity and the Success of Aquatic Invasions. Trends Ecol. Evol. 2007, 22, 454–464. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Macel, M.; Wolfe, L.M.; Biere, A. Population Admixture, Biological Invasions and the Balance between Local Adaptation and Inbreeding Depression. Proc. R. Soc. B Biol. Sci. 2011, 278, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Hovick, S.M.; Whitney, K.D. Hybridisation Is Associated with Increased Fecundity and Size in Invasive Taxa: Meta-Analytic Support for the Hybridisation-Invasion Hypothesis. Ecol. Lett. 2014, 17, 1464–1477. [Google Scholar] [CrossRef] [Green Version]
- Bock, D.G.; Caseys, C.; Cousens, R.D.; Hahn, M.A.; Heredia, S.M.; Huebner, S.; Turner, K.G.; Whitney, K.D.; Rieseberg, L.H. What We Still Don’t Know about Invasion Genetics. Mol. Ecol. 2015, 24, 2277–2297. [Google Scholar] [CrossRef]
- Heckwold, M.J.; Morim, T.; Riccioli, F.; Baltazar-Soares, M. Fresh Start after Rough Rides: Understanding Patterns of Genetic Differentiation upon Human-Mediated Translocations. Biol. Invasions 2021, 23, 3625–3639. [Google Scholar] [CrossRef]
- Stelkens, R.; Seehausen, O. Genetic Distance between Species Predicts Novel Trait Expression in Their Hybrids. Evolution 2009, 63, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Nolte, A.W.; Tautz, D. Understanding the Onset of Hybrid Speciation. Trends Genet. 2010, 26, 54–58. [Google Scholar] [CrossRef]
- Salmenkova, E.A. Population Genetic Processes in Introduction of Fish. Russ. J. Genet. 2008, 44, 758–766. [Google Scholar] [CrossRef]
- Rius, M.; Darling, J.A. How Important Is Intraspecific Genetic Admixture to the Success of Colonising Populations? Trends Ecol. Evol. 2014, 29, 233–242. [Google Scholar] [CrossRef]
- Lavergne, S.; Molofsky, J. Increased Genetic Variation and Evolutionary Potential Drive the Success of an Invasive Grass. Proc. Natl. Acad. Sci. USA 2007, 104, 3883–3888. [Google Scholar] [CrossRef] [Green Version]
- Hathaway, L.; Andersson, S.; Prentice, H.C. Experimental Crosses within European Silene latifolia (Caryophyllaceae): Intraspecific Differentiation, Distance Effects, and Sex Ratio. Botany 2009, 87, 231–240. [Google Scholar] [CrossRef]
- Barker, B.S.; Cocio, J.E.; Anderson, S.R.; Braasch, J.E.; Cang, F.A.; Gillette, H.D.; Dlugosch, K.M. Potential Limits to the Benefits of Admixture during Biological Invasion. Mol. Ecol. 2019, 28, 100–113. [Google Scholar] [CrossRef] [Green Version]
- Facon, B.; Pointier, J.-P.; Jarne, P.; Sarda, V.; David, P. High Genetic Variance in Life-History Strategies within Invasive Populations by Way of Multiple Introductions. Curr. Biol. 2008, 18, 363–367. [Google Scholar] [CrossRef]
- Ebbs, E.T.; Loker, E.S.; Brant, S.V. Phylogeography and Genetics of the Globally Invasive Snail Physa acuta Draparnaud 1805, and Its Potential to Serve as an Intermediate Host to Larval Digenetic Trematodes. BMC Evol. Biol. 2018, 18, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usio, N.; Azuma, N.; Larson, E.R.; Abbott, C.L.; Olden, J.D.; Akanuma, H.; Takamura, K.; Takamura, N. Phylogeographic Insights into the Invasion History and Secondary Spread of the Signal Crayfish in Japan. Ecol. Evol. 2016, 6, 5366–5382. [Google Scholar] [CrossRef] [Green Version]
- Flucher, S.M.; Krapf, P.; Arthofer, W.; Suarez, A.V.; Crozier, R.H.; Steiner, F.M.; Schlick-Steiner, B.C. Effect of Social Structure and Introduction History on Genetic Diversity and Differentiation. Mol. Ecol. 2021, 30, 2511–2527. [Google Scholar] [CrossRef]
- Kolbe, J.J.; Glor, R.E.; Schettino, L.R.; Lara, A.C.; Larson, A.; Losos, J.B. Genetic Variation Increases duringbiological Invasion by a Cuban Lizard. Nature 2004, 431, 177–181. [Google Scholar] [CrossRef]
- Kolbe, J.J.; Larson, A.; Losos, J.B. Differential Admixture Shapes Morphological Variation among Invasive Populations of the Lizard Anolis sagrei. Mol. Ecol. 2007, 16, 1579–1591. [Google Scholar] [CrossRef]
- Schulte, U.; Veith, M.; Mingo, V.; Modica, C.; Hochkirch, A. Strong Genetic Differentiation Due to Multiple Founder Events during a Recent Range Expansion of an Introduced Wall Lizard Population. Biol. Invasions 2013, 15, 2639–2649. [Google Scholar] [CrossRef]
- Zalewski, A.; Michalska-Parda, A.; Ratkiewicz, M.; Kozakiewicz, M.; Bartoszewicz, M.; Brzeziński, M. High Mitochondrial DNA Diversity of an Introduced Alien Carnivore: Comparison of Feral and Ranch American Mink Neovison vison in Poland. Divers. Distrib. 2011, 17, 757–768. [Google Scholar] [CrossRef]
- Frantz, A.C.; Heddergott, M.; Lang, J.; Schulze, C.; Ansorge, H.; Runge, M.; Braune, S.; Michler, F.-U.; Wittstatt, U.; Hoffmann, L.; et al. Limited Mitochondrial DNA Diversity Is Indicative of a Small Number of Founders of the German Raccoon (Procyon lotor) Population. Eur. J. Wildl. Res. 2013, 59, 665–674. [Google Scholar] [CrossRef]
- Gaubert, P.; Del Cerro, I.; Centeno-Cuadros, A.; Palomares, F.; Fournier, P.; Fonseca, C.; Paillat, J.-P.; Godoy, J.A. Tracing Historical Introductions in the Mediterranean Basin: The Success Story of the Common Genet (Genetta genetta) in Europe. Biol. Invasions 2015, 17, 1897–1913. [Google Scholar] [CrossRef] [Green Version]
- Searle, J.B.; Jamieson, P.M.; Gündüz, İ.; Stevens, M.I.; Jones, E.P.; Gemmill, C.E.C.; King, C.M. The Diverse Origins of New Zealand House Mice. Proc. R. Soc. B Biol. Sci. 2009, 276, 209–217. [Google Scholar] [CrossRef] [Green Version]
- King, C.; Alexander, A.; Chubb, T.; Cursons, R.; MacKay, J.; McCormick, H.; Murphy, E.; Veale, A.; Zhang, H. What Can the Geographic Distribution of MtDNA Haplotypes Tell Us about the Invasion of New Zealand by House Mice Mus musculus? Biol. Invasions 2016, 18, 1551–1565. [Google Scholar] [CrossRef]
- Grossen, C.; Biebach, I.; Angelone-Alasaad, S.; Keller, L.F.; Croll, D. Population Genomics Analyses of European Ibex Species Show Lower Diversity and Higher Inbreeding in Reintroduced Populations. Evol. Appl. 2018, 11, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Stipoljev, S.; Safner, T.; Gančević, P.; Galov, A.; Stuhne, T.; Svetličić, I.; Grignolio, S.; Cassinello, J.; Šprem, N. Population Structure and Genetic Diversity of Non-Native Aoudad Populations. Sci. Rep. 2021, 11, 12300. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.B. The Genetics of Mammalian Invasions: A Review. Wildl. Res. 2008, 35, 185. [Google Scholar] [CrossRef]
- Gordon, I.J.; Hester, A.J.; Festa-Bianchet, M.; Gordon, I.J.; Hester, A.J.; Festa-Bianchet, M. Review: The Management of Wild Large Herbivores to Meet Economic, Conservation and Environmental Objectives. J. Appl. Ecol. 2004, 41, 1021–1031. [Google Scholar] [CrossRef]
- Murray, B.D.; Webster, C.R.; Bump, J.K. Broadening the Ecological Context of Ungulate–Ecosystem Interactions: The Importance of Space, Seasonality, and Nitrogen. Ecology 2013, 94, 1317–1326. [Google Scholar] [CrossRef]
- Gawel, A.M.; Rogers, H.S.; Miller, R.H.; Kerr, A.M. Contrasting Ecological Roles of Non-Native Ungulates in a Novel Ecosystem. R. Soc. Open Sci. 2018, 5, 170151. [Google Scholar] [CrossRef] [Green Version]
- Iacolina, L.; Corlatti, L.; Buzan, E.; Safner, T.; Šprem, N. Hybridisation in European Ungulates: An Overview of the Current Status, Causes, and Consequences. Mamm. Rev. 2019, 49, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Hogg, J.T.; Forbes, S.H.; Steele, B.M.; Luikart, G. Genetic Rescue of an Insular Population of Large Mammals. Proc. R. Soc. B Biol. Sci. 2006, 273, 1491–1499. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.M.; Poissant, J.; Hogg, J.T.; Coltman, D.W. Genomic Consequences of Genetic Rescue in an Insular Population of Bighorn Sheep (Ovis canadensis). Mol. Ecol. 2012, 21, 1583–1596. [Google Scholar] [CrossRef]
- Gompert, Z. Population Genomics as a New Tool for Wildlife Management. Mol. Ecol. 2012, 21, 1542–1544. [Google Scholar] [CrossRef]
- Giglio, R.M.; Ivy, J.A.; Jones, L.C.; Latch, E.K. Evaluation of Alternative Management Strategies for Maintenance of Genetic Variation in Wildlife Populations. Anim. Conserv. 2016, 19, 380–390. [Google Scholar] [CrossRef]
- Apollonio, M.; Belkin, V.V.; Borkowski, J.; Borodin, O.I.; Borowik, T.; Cagnacci, F.; Danilkin, A.A.; Danilov, P.I.; Faybich, A.; Ferretti, F.; et al. Challenges and Science-Based Implications for Modern Management and Conservation of European Ungulate Populations. Mammal Res. 2017, 62, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Ballari, S.A.; Cuevas, M.F.; Cirignoli, S.; Valenzuela, A.E.J. Invasive Wild Boar in Argentina: Using Protected Areas as a Research Platform to Determine Distribution, Impacts and Management. Biol. Invasions 2015, 17, 1595–1602. [Google Scholar] [CrossRef]
- Aschim, R.A.; Brook, R.K. Evaluating Cost-Effective Methods for Rapid and Repeatable National Scale Detection and Mapping of Invasive Species Spread. Sci. Rep. 2019, 9, 7254. [Google Scholar] [CrossRef]
- Markov, N.; Economov, A.; Hjeljord, O.; Rolandsen, C.M.; Bergqvist, G.; Danilov, P.; Dolinin, V.; Kambalin, V.; Kondratov, A.; Krasnoshapka, N.; et al. The Wild Boar Sus scrofa in Northern Eurasia: A Review of Range Expansion History, Current Distribution, Factors Affecting the Northern Distributional Limit, and Management Strategies. Mamm. Rev. 2022, 52, 519–537. [Google Scholar] [CrossRef]
- Massei, G.; Kindberg, J.; Licoppe, A.; Gačić, D.; Šprem, N.; Kamler, J.J.; Baubet, E.; Hohmann, U.; Monaco, A.; Ozoliņš, J.; et al. Wild Boar Populations up, Numbers of Hunters down? A Review of Trends and Implications for Europe. Pest Manag. Sci. 2015, 71, 492–500. [Google Scholar] [CrossRef]
- Keuling, O.; Podgórski, T.; Monaco, A.; Meletti, M.; Merta, D.; Albrycht, M.; Genov, P.V.; Gethöffer, F.; Vetter, S.G.; Jori, F.; et al. Eurasian Wild Boar Sus scrofa (Linnaeus, 1758). In Ecology, Conservation and Management of Wild Pigs and Peccaries; Meletti, M., Meijaard, E., Eds.; Cambridge University Press: New York, NY, USA, 2018; pp. 202–233. [Google Scholar]
- Risch, D.R.; Ringma, J.; Price, M.R. The Global Impact of Wild Pigs (Sus scrofa) on Terrestrial Biodiversity. Sci. Rep. 2021, 11, 13256. [Google Scholar] [CrossRef]
- García, G.; Vergara, J.; Lombardi, R. Genetic Characterization and Phylogeography of the Wild Boar Sus scrofa Introduced into Uruguay. Genet. Mol. Biol. 2011, 34, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.S.; Corn, J.L.; Mayer, J.J.; Jordan, T.R.; Farnsworth, M.L.; Burdett, C.L.; VerCauteren, K.C.; Sweeney, S.J.; Miller, R.S. Historical, Current, and Potential Population Size Estimates of Invasive Wild Pigs (Sus scrofa) in the United States. Biol. Invasions 2019, 21, 2373–2384. [Google Scholar] [CrossRef]
- Wehr, N.H. Historical Range Expansion and Biological Changes of Sus scrofa Corresponding to Domestication and Feralization. Mammal Res. 2021, 66, 1–12. [Google Scholar] [CrossRef]
- Bengsen, A.J.; West, P.; Krull, C.R. Feral Pigs in Australia and New Zealand: Range, Trend, Management and Impacts of an Invasive SpeciesTitle. In Ecology, Conservation and Management of Wild Pigs and Peccaries; Meletti, M., Meijaard, E., Eds.; Cambridge University Press: New York, NY, USA, 2018; pp. 325–338. ISBN 978-1-107-18731-3. [Google Scholar]
- Danilkin, A. Pigs (Suidae); GEOS: Moscow, Russia, 2002; ISBN 5-89118-228-09. [Google Scholar]
- Tikhonov, V.N.; Knyazev, S.P. Immunogenetic Traits in Some Forms of Wild Pigs of Europe, Asia, Africa and America. In Morphology and Genetics of Wild Boar; Davletova, L.V., Ed.; Nauka: Moscow, Russia, 1985; pp. 3–16. [Google Scholar]
- Larson, G.; Dobney, K.; Albarella, U.; Fang, M.; Matisoo-Smith, E.; Robins, J.; Lowden, S.; Finlayson, H.; Brand, T.; Willerslev, E.; et al. Worldwide Phylogeography of Wild Boar Reveals Multiple Centers of Pig Domestication. Science 2005, 307, 1618–1621. [Google Scholar] [CrossRef] [Green Version]
- Ashrafzadeh, M.R.; Rezaei, H.R.; Khalilipour, O.; Kusza, S. Genetic Relationships of Wild Boars Highlight the Importance of Southern Iran in Forming a Comprehensive Picture of the Species’ Phylogeography. Mamm. Biol. 2018, 92, 21–29. [Google Scholar] [CrossRef]
- Ruvinsky, A.; Rothschild, M.F.; Larson, G.; Gongora, J. Systematics and Evolution the Pig. In The Genetics of the Pig; Rothschild, M.F., Ruvinsky, A., Eds.; CAB International: Oxforshire, UK, 2011; pp. 1–13. [Google Scholar]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of Pig Genomes Provide Insight into Porcine Demography and Evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Alexandri, P.; Triantafyllidis, A.; Papakostas, S.; Chatzinikos, E.; Platis, P.; Papageorgiou, N.; Larson, G.; Abatzopoulos, T.J.; Triantaphyllidis, C. The Balkans and the Colonization of Europe: The Post-Glacial Range Expansion of the Wild Boar, Sus scrofa. J. Biogeogr. 2012, 39, 713–723. [Google Scholar] [CrossRef]
- Kusza, S.; Podgórski, T.; Scandura, M.; Borowik, T.; Jávor, A.; Sidorovich, V.E.; Bunevich, A.N.; Kolesnikov, M.; Jȩdrzejewska, B. Contemporary Genetic Structure, Phylogeography and Past Demographic Processes of Wild Boar Sus scrofa Population in Central and Eastern Europe. PLoS ONE 2014, 9, e91401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilaça, S.T.; Biosa, D.; Zachos, F.; Iacolina, L.; Kirschning, J.; Alves, P.C.; Paule, L.; Gortazar, C.; Mamuris, Z.; Jędrzejewska, B.; et al. Mitochondrial Phylogeography of the European Wild Boar: The Effect of Climate on Genetic Diversity and Spatial Lineage Sorting across Europe. J. Biogeogr. 2014, 41, 987–998. [Google Scholar] [CrossRef]
- Veličković, N.; Djan, M.; Ferreira, E.; Stergar, M.; Obreht, D.; Maletić, V.; Fonseca, C. From North to South and Back: The Role of the Balkans and Other Southern Peninsulas in the Recolonization of Europe by Wild Boar. J. Biogeogr. 2015, 42, 716–728. [Google Scholar] [CrossRef]
- Niedziałkowska, M.; Tarnowska, E.; Ligmanowska, J.; Jędrzejewska, B.; Podgórski, T.; Radziszewska, A.; Ratajczyk, I.; Kusza, S.; Bunevich, A.N.; Danila, G.; et al. Clear Phylogeographic Pattern and Genetic Structure of Wild Boar Sus scrofa Population in Central and Eastern Europe. Sci. Rep. 2021, 11, 9680. [Google Scholar] [CrossRef]
- Watanobe, T.; Ishiguro, N.; Nakano, M. Phylogeography and Population Structure of the Japanese Wild Boar Sus scrofa Leucomystax: Mitochondrial DNA Variation. Zoolog. Sci. 2003, 20, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.C.; Han, S.H.; Fang, M.; Lee, S.S.; Ko, M.S.; Lee, H.; Lim, H.T.; Yoo, C.K.; Lee, J.H.; Jeon, J.T. The Robust Phylogeny of Korean Wild Boar (Sus scrofa soreanus) Using Partial D-Loop Sequence of MtDNA. Mol. Cells 2009, 28, 423–430. [Google Scholar] [CrossRef]
- Ramayo, Y.; Shemeret’eva, I.N.; Pérez-Enciso, M. Mitochondrial DNA Diversity in Wild Boar from the Primorsky Krai Region (East Russia). Anim. Genet. 2011, 42, 96–99. [Google Scholar] [CrossRef]
- Choi, S.K.; Lee, J.-E.; Kim, Y.-J.; Min, M.-S.; Voloshina, I.; Myslenkov, A.; Oh, J.G.; Kim, T.-H.; Markov, N.; Seryodkin, I.; et al. Genetic Structure of Wild Boar (Sus scrofa) Populations from East Asia Based on Microsatellite Loci Analyses. BMC Genet. 2014, 15, 85. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.K.; Kim, K.S.; Ranyuk, M.; Babaev, E.; Voloshina, I.; Bayarlkhagva, D.; Chong, J.R.; Ishiguro, N.; Yu, L.; Min, M.S.; et al. Asia-Wide Phylogeography of Wild Boar (Sus scrofa) Based on Mitochondrial DNA and Y-Chromosome: Revising the Migration Routes of Wild Boar in Asia. PLoS ONE 2020, 15, e0238049. [Google Scholar] [CrossRef]
- Gongora, J.; Fleming, P.; Spencer, P.B.S.; Mason, R.; Garkavenko, O.; Meyer, J.-N.; Droegemueller, C.; Lee, J.H.; Moran, C. Phylogenetic Relationships of Australian and New Zealand Feral Pigs Assessed by Mitochondrial Control Region Sequence and Nuclear GPIP Genotype. Mol. Phylogenet. Evol. 2004, 33, 339–348. [Google Scholar] [CrossRef]
- Burgos-Paz, W.; Souza, C.A.; Megens, H.J.; Ramayo-Caldas, Y.; Melo, M.; Lemús-Flores, C.; Caal, E.; Soto, H.W.; Martínez, R.; Álvarez, L.A.; et al. Porcine Colonization of the Americas: A 60k SNP Story. Heredity 2013, 110, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, B.E.; Malek, M.J.; Newman, R.A.; Schmit, B.S.; Swafford, S.R.; Sweitzer, R.A.; Simmons, R.B. Mitochondrial Diversity Supports Multiple Origins for Invasive Pigs. J. Wildl. Manage. 2014, 78, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Zinovieva, N.A.; Kostyunina, O.V.; Ekonomov, A.V.; Shevnina, M.S.; Domskij, I.A.; Gladyr’, E.A.; Brem, G. Polymorphism of genes assocciated with the quantitative trait loci in wild boar (Sus scrofa L., 1758) in Russia. Sel’skokhozyaistvennaya Biol. 2013, 100, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Davidova, E.E.; Varnakov, A.P.; Dankvert, S.A.; Soltynskaya, I.V.; Seliverstova, A.S.; Ignatova, I.A.; Tsvetkov, I.A.; Pronyaev, A.V.; Davidov, A.V.; Vovchenko, V.E.; et al. The Molecular Genetic Differentiation of Subspecies and Other Forms of Wild Boar (Sus scrofa L.) in the Territory of Russia. Vestn. Okhotovedenia 2013, 4, 44–52. [Google Scholar]
- Kostyunina, O.; Traspov, A.; Economov, A.; Seryodkin, I.; Senchik, A.; Bakoev, N.; Prytkov, Y.; Bardukov, N.; Domsky, I.; Karpushkina, T. Genetic Diversity, Admixture and Analysis of Homozygous-by-Descent (HBD) Segments of Russian Wild Boar. Biology 2022, 11, 203. [Google Scholar] [CrossRef]
- Heptner, V.G.; Naumov, N.P.; Yurgenson, P.B.; Sludskii, A.A.; Chirkova, A.F.; Bannikov, A.G. Mammals of the Soviet Union. Vol.1—Artiodactyla and Perissodactyla; Vishaya Shkola: Moscow, Russia, 1961. [Google Scholar]
- Genov, P.; Nikolovg, H.; Massei, G.; Gerasimov, S. Craniometrical Analysis of Bulgarian Wild Boar (Sus scrofa) Populations. J. Zool. 1991, 225, 309–325. [Google Scholar] [CrossRef]
- Keuling, O.; Leus, K. Sus scrofa. The IUCN Red List of Threatened Species 2019: E.T41775A44141833. 2019. Available online: https://www.iucnredlist.org/species/41775/44141833 (accessed on 20 July 2021).
- Giuffra, E.; Kijas, J.M.H.; Amarger, V.; Calborg, O.; Jeon, J.-T.; Andersson, L. The Origin of the Domestic Pig: Independent Domestication and Subsequent Introgression. Genetics 2000, 154, 1785–1791. [Google Scholar] [CrossRef]
- Fernandez, A.I.; Alves, E.; Ovilo, C.; Rodriguez, M.C.; Silio, L. Divergence Time Estimates of East Asian and European Pigs Based on Multiple near Complete Mitochondrial DNA Sequences. Anim. Genet. 2010, 42, 86–88. [Google Scholar] [CrossRef]
- Kiselev, A.A. Wild Boar in the Middle Urals. Okhota i Okhotnichye Khoz-Vo 1986, 12–14. [Google Scholar]
- Markov, N.I. Population Dynamics of Wild Boar, Sus scrofa, in Sverdlovsk Oblast and Its Relation to Climatic Factors. Russ. J. Ecol. 1997, 28, 269–274. [Google Scholar]
- Melis, C.; Szafranska, P.A.; Jedrzejewska, B.; Barton, K. Biogeographical Variation in the Population Density of Wild Boar (Sus scrofa) in Western Eurasia. J. Biogeogr. 2006, 33, 803–811. [Google Scholar] [CrossRef]
- Vetter, S.G.; Ruf, T.; Bieber, C.; Arnold, W. What Is a Mild Winter? Regional Differences in within-Species Responses to Climate Change. PLoS ONE 2015, 10, e0132178. [Google Scholar] [CrossRef] [PubMed]
- Markov, N.; Pankova, N.; Morelle, K. Where Winter Rules: Modeling Wild Boar Distribution in Its North-Eastern Range. Sci. Total Environ. 2019, 687, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Khederzadeh, S.; Kusza, S.; Huang, C.P.; Markov, N.; Scandura, M.; Babaev, E.; Šprem, N.; Seryodkin, I.V.; Paule, L.; Esmailizadeh, A.; et al. Maternal Genomic Variability of the Wild Boar (Sus scrofa) Reveals the Uniqueness of East-Caucasian and Central Italian Populations. Ecol. Evol. 2019, 9, 9467–9478. [Google Scholar] [CrossRef] [Green Version]
- Okumura, N.; Ishiguro, N.; Nakano, M.; Hirai, K.; Matsui, A.; Sahara, M. Geographic Population Structure and Sequence Divergence in the Mitochondrial DNA Control Region of the Japanese Wild Boar (Sus scrofa leucomystax), with Reference to Those of Domestic Pigs. Biochem. Genet. 1996, 34, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Watanobe, T.; Ishiguro, N.; Okumura, N.; Nakano, M.; Matsui, A.; Hongo, H.; Ushiro, H. Ancient Mitochondrial DNA Reveals the Origin of Sus scrofa from Rebun Island, Japan. J. Mol. Evol. 2001, 52, 281–289. [Google Scholar] [CrossRef]
- Khalilzadeh, P.; Rezaei, H.R.; Fadakar, D.; Serati, M.; Aliabadian, M.; Haile, J.; Goshtasb, H. Contact Zone of Asian and European Wild Boar at North West of Iran. PLoS ONE 2016, 11, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Kijas, J.M.H.; Andersson, L. A Phylogenetic Study of the Origin of the Domestic Pig Estimated from the Near-Complete MtDNA Genome. J. Mol. Evol. 2001, 52, 302–308. [Google Scholar] [CrossRef]
- Fitzpatrick, B.M.; Fordyce, J.A.; Niemiller, M.L.; Reynolds, R.G. What Can DNA Tell Us about Biological Invasions? Biol. Invasions 2012, 14, 245–253. [Google Scholar] [CrossRef]
- Lin, C.-S.; Sun, Y.-L.; Liu, C.-Y.; Yang, P.-C.; Chang, L.-C.; Cheng, I.-C.; Mao, S.J.T.; Huang, M.-C. Complete Nucleotide Sequence of Pig (Sus scrofa) Mitochondrial Genome and Dating Evolutionary Divergence within Artiodactyla. Gene 1999, 236, 107–114. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J Mol. Evol 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, P.; Pinheiro, I.; Godinho, R.; Vicente, J.; Gortázar, C.; Scandura, M. Genetic Diversity of Wild Boar Populations and Domestic Pig Breeds (Sus scrofa) in South-Western Europe. Biol. J. Linn. Soc. 2010, 101, 797–822. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree Version 1.3.1. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 16 January 2020).
- Paradis, E. Pegas: An R Package for Population Genetics with an Integrated-Modular Approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A Cladistic Analysis of Phenotypic Associations with Haplotypes Inferred from Restriction Endonuclease Mapping and DNA Sequence Data. III. Cladogram Estimation. Genetics 1992, 132, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Gogin, A.; Gerasimov, V.; Malogolovkin, A.; Kolbasov, D. African Swine Fever in the North Caucasus Region and the Russian Federation in Years 2007–2012. Virus Res. 2013, 173, 198–203. [Google Scholar] [CrossRef]
- Scandura, M.; Iacolina, L.; Crestanello, B.; Pecchioli, E.; Di Benedetto, M.F.; Russo, V.; Davoli, R.; Apollonio, M.; Bertorelle, G. Ancient vs. Recent Processes as Factors Shaping the Genetic Variation of the European Wild Boar: Are the Effects of the Last Glaciation Still Detectable? Mol. Ecol. 2008, 17, 1745–1762. [Google Scholar] [CrossRef]
- Sagua, M.I.; Figueroa, C.E.; Acosta, D.B.; Fernández, G.P.; Carpinetti, B.N.; Birochio, D.; Merino, M.L. Inferring the Origin and Genetic Diversity of the Introduced Wild Boar (Sus scrofa) Populations in Argentina: An Approach from Mitochondrial Markers. Mammal Res. 2018, 63, 467–476. [Google Scholar] [CrossRef]
- Albarella, U.; Dobney, K.; Rowley-Conwy, P. Size and Shape of the Eurasian Wild Boar ( Sus scrofa ), with a View to the Reconstruction of Its Holocene History. Environ. Archaeol. 2009, 14, 103–136. [Google Scholar] [CrossRef]
- Wu, C.Y.; Jiang, Y.N.; Chu, H.P.; Li, S.H.; Wang, Y.; Li, Y.H.; Chang, Y.; Ju, Y.T. The Type I Lanyu Pig Has a Maternal Genetic Lineage Distinct from Asian and European Pigs. Anim. Genet. 2007, 38, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.V.; Brandebourg, T.D.; Kohn, M.C.; Đikić, D.; Irwin, M.H.; Pinkert, C.A. Mitochondrial DNA Sequence and Phylogenetic Evaluation of Geographically Disparate Sus Scrofa Breeds. Anim. Biotechnol. 2015, 26, 17–28. [Google Scholar] [CrossRef]
- Goedbloed, D.J.; Megens, H.J.; Van Hooft, P.; Herrero-Medrano, J.M.; Lutz, W.; Alexandri, P.; Crooijmans, R.P.M.A.; Groenen, M.; Van Wieren, S.E.; Ydenberg, R.C.; et al. Genome-Wide Single Nucleotide Polymorphism Analysis Reveals Recent Genetic Introgression from Domestic Pigs into Northwest European Wild Boar Populations. Mol. Ecol. 2013, 22, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Iacolina, L.; Pertoldi, C.; Amills, M.; Kusza, S.; Megens, H.-J.; Bâlteanu, V.A.; Bakan, J.; Cubric-Curik, V.; Oja, R.; Saarma, U.; et al. Hotspots of Recent Hybridization between Pigs and Wild Boars in Europe. Sci. Rep. 2018, 8, 17372. [Google Scholar] [CrossRef] [Green Version]
- Frantz, A.C.; Zachos, F.E.; Kirschning, J.; Cellina, S.; Bertouille, S.; Mamuris, Z.; Koutsogiannouli, E.A.; Burke, T. Genetic Evidence for Introgression between Domestic Pigs and Wild Boars (Sus scrofa) in Belgium and Luxembourg: A Comparative Approach with Multiple Marker Systems. Biol. J. Linn. Soc. 2013, 110, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.M.; Stamps, J.A. The Effect of Natal Experience on Habitat Preferences. Trends Ecol. Evol. 2004, 19, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Bonte, D.; Hovestadt, T.; Poethke, H. Evolution of Dispersal Polymorphism and Local Adaptation of Dispersal Distance in Spatially Structured Landscapes. Oikos 2010, 119, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.-S.S.; Rajasekar, S.; St. John, J.C. The Relationship between Mitochondrial DNA Haplotype and the Reproductive Capacity of Domestic Pigs (Sus scrofa domesticus). BMC Genet. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Mary, N.; Iannuccelli, N.; Petit, G.; Bonnet, N.; Pinton, A.; Barasc, H.; Amélie, F.; Calgaro, A.; Grosbois, V.; Servin, B.; et al. Genome-wide Analysis of Hybridization in Wild Boar Populations Reveals Adaptive Introgression from Domestic Pig. Evol. Appl. 2022, 15, 1115–1128. [Google Scholar] [CrossRef]
- Hewitt, G.M. Post-Glacial Re-Colonization of European Biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.; Cosson, J. Comparative Phylogeography and Postglacial Colonization Routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, R.S.; Zachos, F.E. Fossil Evidence and Phylogeography of Temperate Species: ‘Glacial Refugia’ and Post-Glacial Recolonization. J. Biogeogr. 2009, 36, 2013–2020. [Google Scholar] [CrossRef]
- Meiri, M.; Huchon, D.; Bar-Oz, G.; Boaretto, E.; Horwitz, L.K.; Maeir, A.M.; Sapir-Hen, L.; Larson, G.; Weiner, S.; Finkelstein, I. Ancient DNA and Population Turnover in Southern Levantine Pigs—Signature of the Sea Peoples Migration? Sci. Rep. 2013, 3, 3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, K.M.; Nichols, R.A.; Hewitt, G.M. Spatial Patterns of Genetic Variation Generated by Different Forms of Dispersal during Range Expansion. Heredity 1996, 77, 282–291. [Google Scholar] [CrossRef]
- Hewitt, G. The Genetic Legacy of the Quaternary Ice Ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Geographical Region | n | H | D-loop Haplotypes 637-bp (Number of Individuals) * | Private Haplotypes ** |
|---|---|---|---|---|
| Urals | 116 | 11 | Ss1(35), Ss2(57), Ss3(6), Ss4(2), Ss5(1), Ss6(3), Ss7(4), Ss8(1), Ss9(4), Ss13(2), Ss14(1) | Ss4, Ss5, Ss8, Ss13 |
| Caucasus (Republic of Dagestan) | 26 | 3 | Ss1(11), Ss7 (14), Ss11(1) | Ss11 |
| Central Asia (Kazakhstan and Uzbekistan) | 16 | 4 | Ss6 (8), Ss9 (1), Ss10(6), Ss12 (1), | Ss10, Ss12 |
| Russian Far East | 38 | 7 | Ss15(4), Ss16(20), Ss17(1), Ss18(4), Ss19(4), Ss20(2), Ss21(3) | Ss15-21 |
| Total | 196 |
| Location | Hd | π (%) | Tajima’s D | Fu’s Fs | Source |
|---|---|---|---|---|---|
| Urals | 0.667 ± 0.033 (0.649 ± 0.034) a | 0.00657 ± 0.0011 (0.0059 ± 0.0015) a | −0.78 (−0.77 a) n. s. | 2.21 * (3.061 a**) | This study |
| Caucasus (Republic of Dagestan) | 0.551 ± 0.048 | 0.0099 ± 0.0009 | 2.42 *** | 10.65 *** | This study |
| Central Asia (Kazakhstan and Uzbekistan) | 0.642 ± 0.081 | 0.0024 ± 0.001 | −1.6* | 0.718n. s. | This study |
| Russian Far East | 0.698 ± 0.072 (0.682 ± 0.074 a) | 0.0061 ± 0.001 (0.00514 ± 0.001 a) | −0.77(0.72 a) n. s. | 2.36 (2.53) *. | This study |
| Central and Eastern Europe | 0.65 b | 0.00570 ± 0.0015 b | −1.503 c, n. s. | −5.051 c, n. s. | Niedziałkowska et al. 2021 |
| Year of Release | Number of Individuals | Origin | Clade |
|---|---|---|---|
| 1978 | 30 | Central Asia (Kyrgyzstan) | Eastern |
| 1979 | 171 | Belarus (Brest region) and Caucasus (Kabardino-Balkaria) | Western |
| 1981 | 123 | Far East of Russia (Primorsky Krai) | Eastern |
| 1982 | 94 | Western Russia (Smolensk Oblast) | Mixed (67% Western, 33% Eastern) * |
| 1983 | 101 | Western Russia (Smolensk Oblast) | Mixed (67% Western, 33% Eastern) * |
| 1984 | 97 | Western Russia (Kaluga Oblast) | Mixed (89% Western, 11% Eastern) * |
| 1984 | 50 | Western Russia (Vladimir Oblast) | European* |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markov, N.I.; Ranyuk, M.N.; Babaev, E.A.; Seryodkin, I.V.; Senchik, A.V.; Bykova, E.A.; Esipov, A.V.; Nurtazin, S.T.; Pavlova, O.S.; Matrosova, V.A. Introduced, Mixed, and Peripheral: Conservation of Mitochondrial-DNA Lineages in the Wild Boar (Sus scrofa L.) Population in the Urals. Diversity 2022, 14, 916. https://doi.org/10.3390/d14110916
Markov NI, Ranyuk MN, Babaev EA, Seryodkin IV, Senchik AV, Bykova EA, Esipov AV, Nurtazin ST, Pavlova OS, Matrosova VA. Introduced, Mixed, and Peripheral: Conservation of Mitochondrial-DNA Lineages in the Wild Boar (Sus scrofa L.) Population in the Urals. Diversity. 2022; 14(11):916. https://doi.org/10.3390/d14110916
Chicago/Turabian StyleMarkov, Nikolay I., Maryana N. Ranyuk, Elmar A. Babaev, Ivan V. Seryodkin, Alexander V. Senchik, Elena A. Bykova, Alexander V. Esipov, Sabir T. Nurtazin, Olga S. Pavlova, and Vera A. Matrosova. 2022. "Introduced, Mixed, and Peripheral: Conservation of Mitochondrial-DNA Lineages in the Wild Boar (Sus scrofa L.) Population in the Urals" Diversity 14, no. 11: 916. https://doi.org/10.3390/d14110916