
Metagenomics Analysis Reveals the Microbial Communities, Antimicrobial Resistance Gene Diversity and Potential Pathogen Transmission Risk of Two Different Landfills in China

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Illumina Sequencing
2.3. Data Analysis
3. Results and Discussion
3.1. Sequence Data
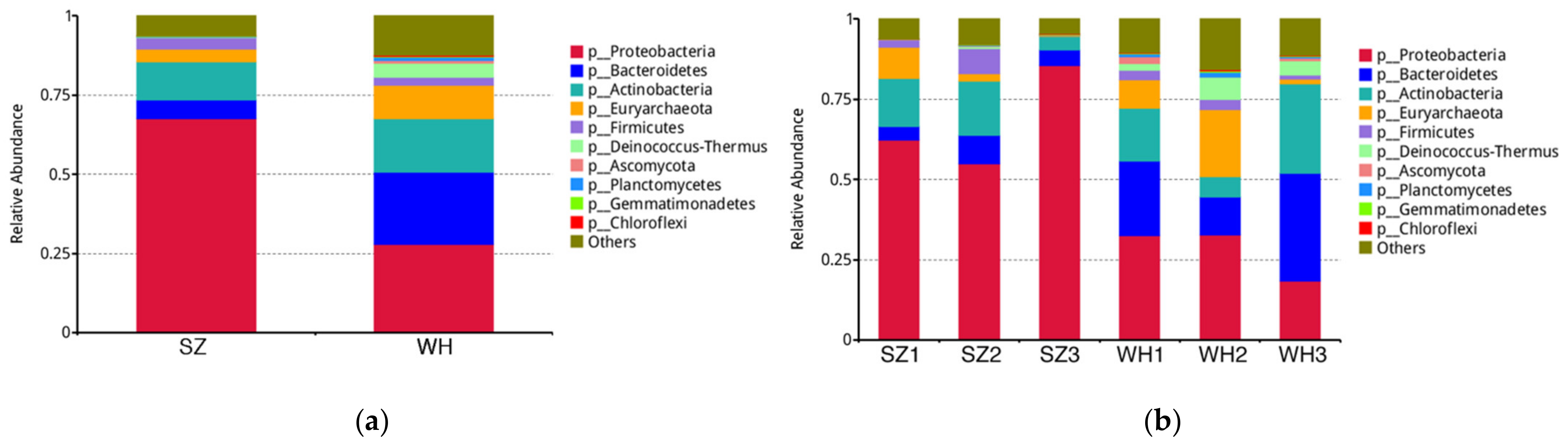
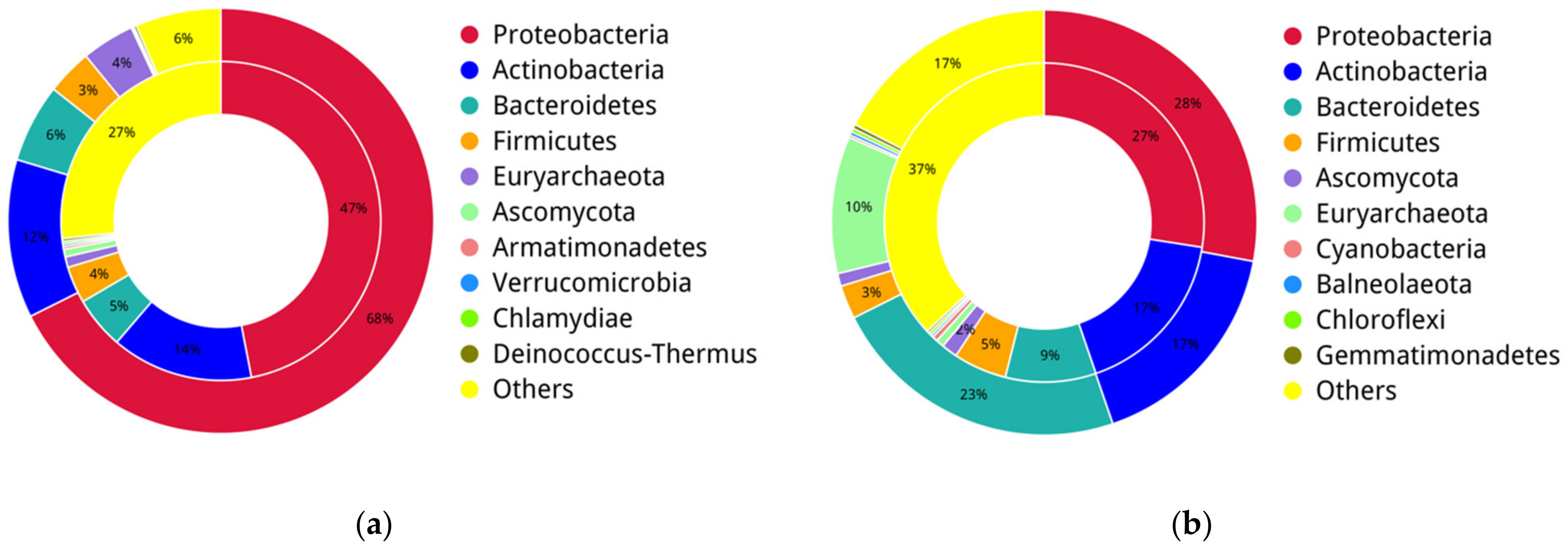
3.2. Microbial Taxonomy Composition at the Phylum Level
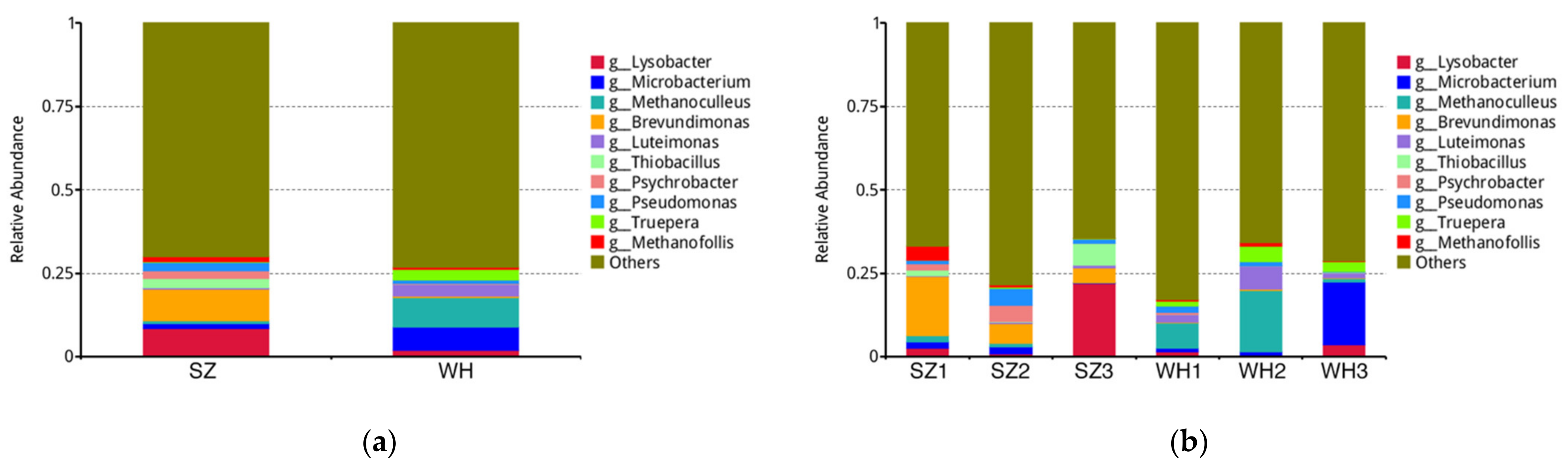
3.3. Microbial Taxonomy Composition at the Genus Level
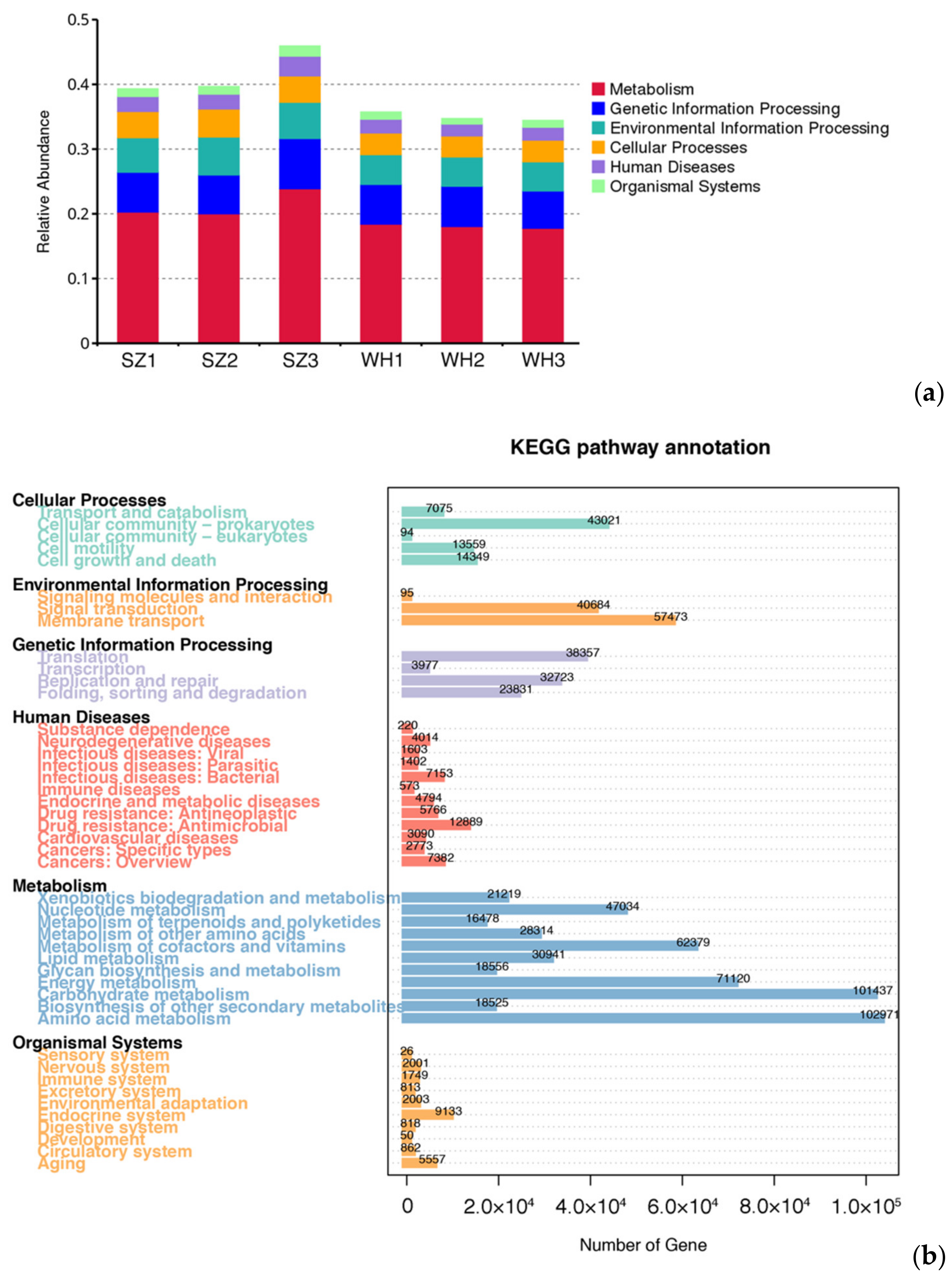
3.4. Gene Function Prediction of the Microbial Communities
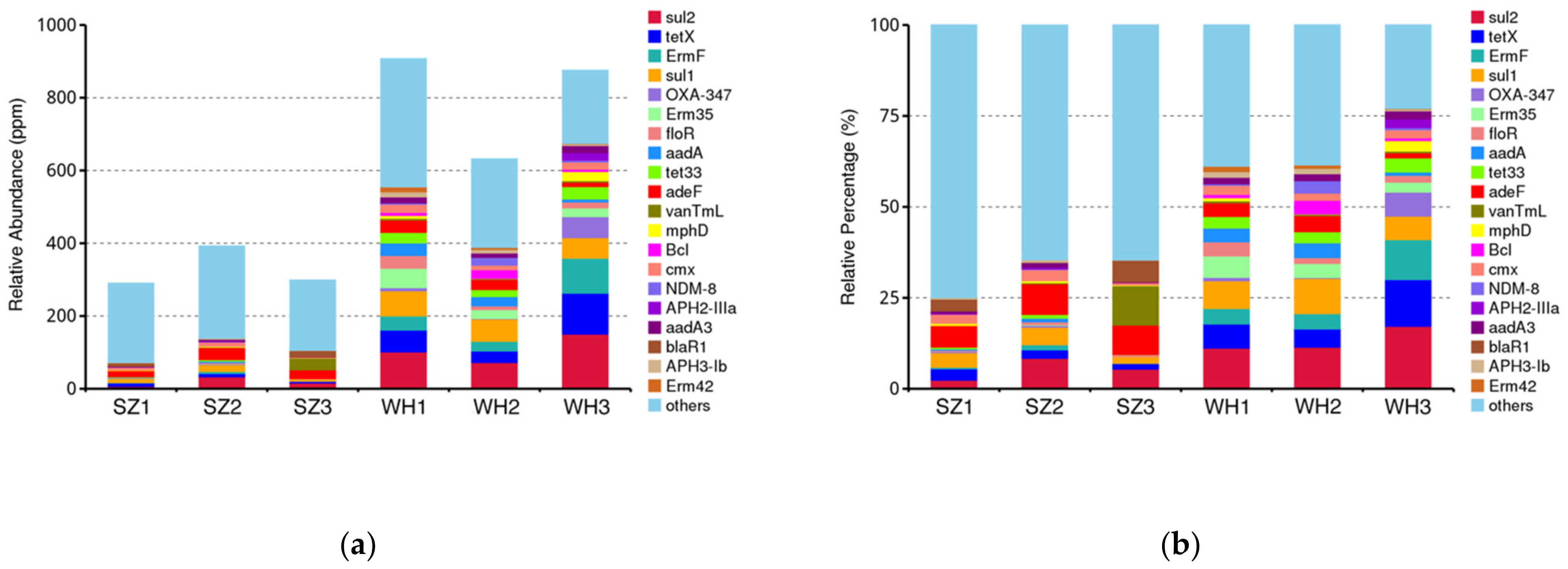
3.5. ARGs Diversity
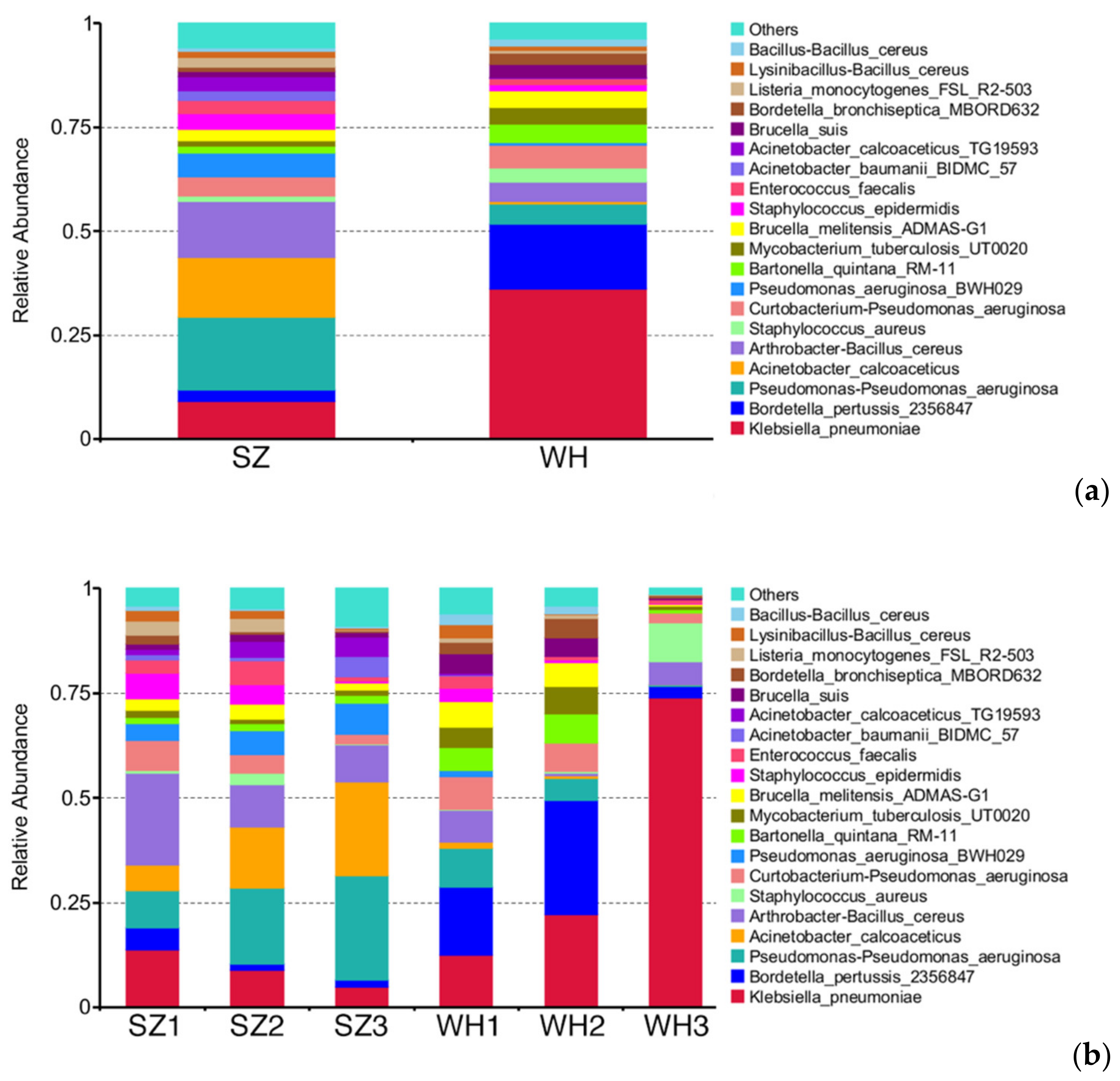
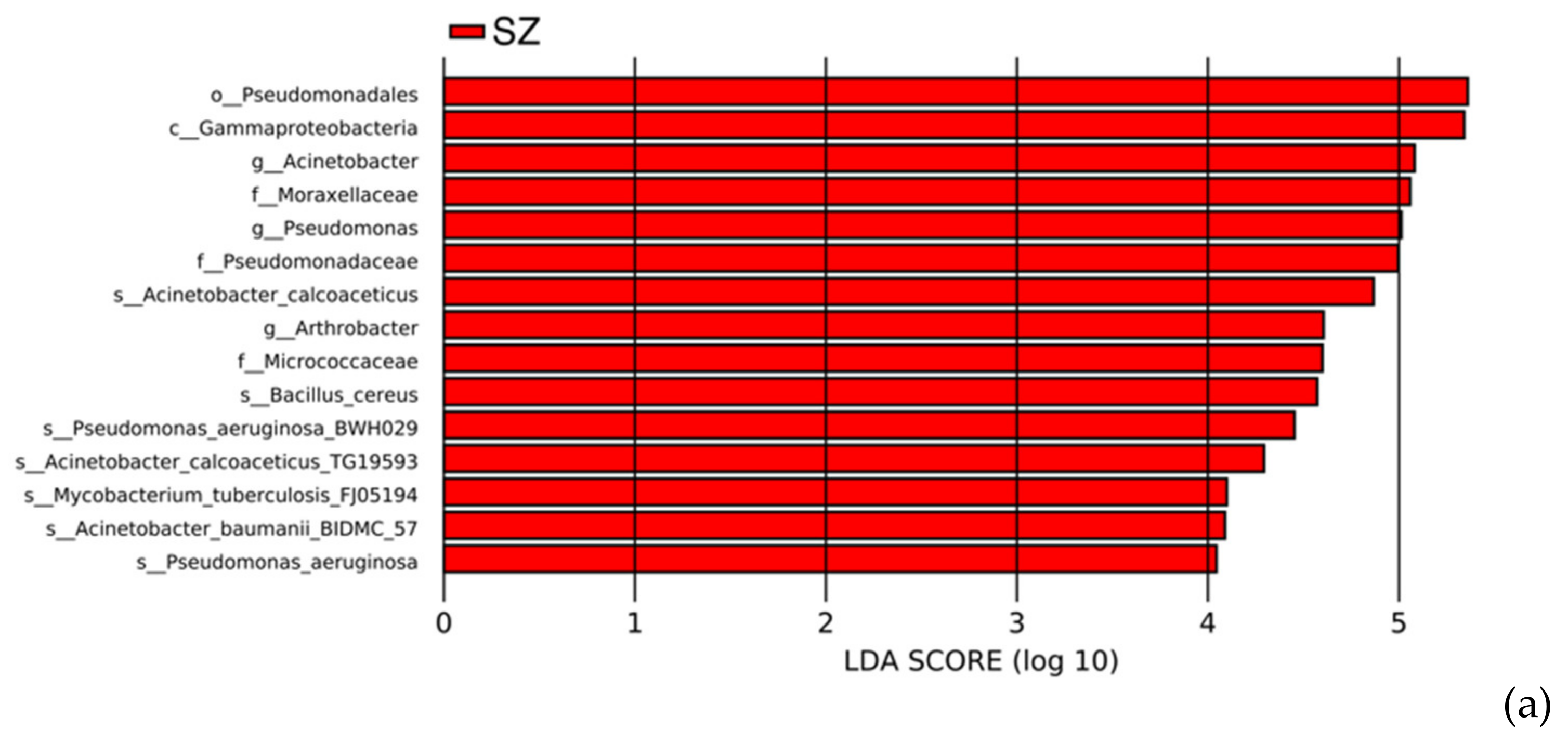
3.6. HPB Species Diversity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sekhohola-Dlamini, L.; Tekere, M. Microbiology of municipal solid waste landfills: A review of microbial dynamics and ecological influences in waste bioprocessing. Biodegradation 2020, 31, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Ecology and Environment of the People’s Republic of China. Statical Yearbook of Prevention and Control of Environmental Pollution by Solid Waste in Big and Medium-Sized Cities; Ministry of Ecology and Environment of the People’s Republic of China: Beijing, China, 2020.
- Ministry of Housing and Urban-Rural Development of the People’s Republic of China (MOHURD). China Urban Rural Construction Statistical Yearbook; Ministry of Housing and Urban-Rural Development of the People’s Republic of China (MOHURD): Beijing, China, 2020.
- Baziene, K.; Tetsman, I.; Albrektiene, R. Level of Pollution on Surrounding Environment from Landfill Aftercare. Int. J. Environ. Res. Public Health 2020, 17, 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, M.; Yoneda, K.; Mohd-Zaki, Z.; Amir, A.; Othman, N. Heavy metals in leachate, impacted soils and natural soils of different landfills in Malaysia: An alarming threat. Chemosphere 2021, 267, 128874. [Google Scholar] [CrossRef] [PubMed]
- Gujre, N.; Mitra, S.; Soni, A.; Agnihotri, R.; Rangan, L.; Rene, E.R.; Sharma, M.P. Speciation, contamination, ecological and human health risks assessment of heavy metals in soils dumped with municipal solid wastes. Chemosphere 2021, 262, 128013. [Google Scholar] [CrossRef]
- Ziyang, L.; Luochun, W.; Nanwen, Z.; Youcai, Z. Martial recycling from renewable landfill and associated risks: A review. Chemosphere 2015, 131, 91–103. [Google Scholar] [CrossRef]
- Wanka, S.; Münnich, K.; Fricke, K. Landfill Mining—Wet mechanical treatment of fine MSW with a wet jigger. Waste Manag. 2017, 59, 316–323. [Google Scholar] [CrossRef]
- Gerba, C.P.; Tamimi, A.H.; Pettigrew, C.; Weisbrod, A.V.; Rajagopalan, V. Sources of microbial pathogens in municipal solid waste landfills in the United States of America. Waste Manag. Res. 2011, 29, 781–790. [Google Scholar] [CrossRef]
- Nair, A.T. Bioaerosols in the landfill environment: An overview of microbial diversity and potential health hazards. Aerobiologia 2021, 4, 1–19. [Google Scholar]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 15175. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- McArthur, A.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Xia, Y.; Li, B.; Yang, Y.; Li, L.G.; Tiedje, J.M.; Zhang, T. Metagenomic Assembly Reveals Hosts of Antibiotic Resistance Genes and the Shared Resistome in Pig, Chicken, and Human Feces. Environ. Sci. Technol. 2016, 50, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wang, H.; Cai, L.; Yu, Y. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ. Sci. Technol. 2015, 49, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Zainun, M.Y.; Simarani, K. Metagenomics profiling for assessing microbial diversity in both active and closed landfills. Sci. Total Environ. 2018, 616–617, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.; Chownk, M.; Kumar, V.; Purohit, A.; Vashisht, A.; Kumar, V.; Yadav, S.K. Bioprospecting potential of microbial communities in solid waste landfills for novel enzymes through metagenomic approach. World J. Microbiol. Biotechnol. 2020, 36, 34. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.K.M.; Sakamoto, I.K.; Varesche, M.B.A.; Wendland, E. Microbial structure and diversity in non-sanitary landfills and association with physicochemical parameters. Environ. Sci. Pollut. Res. Int. 2020, 27, 40690–40705. [Google Scholar] [CrossRef]
- Xie, B.; Xiong, S.; Liang, S.; Hu, C.; Zhang, X.; Lu, J. Performance and bacterial compositions of aged refuse reactors treating mature landfill leachate. Bioresour. Technol. 2012, 103, 71–77. [Google Scholar] [CrossRef]
- Köchling, T.; Sanz, J.L.; Gavazza, S.; Florencio, L. Analysis of microbial community structure and composition in leachates from a young landfill by 454 pyrosequencing. Appl. Microbiol. Biotechnol. 2015, 99, 5657–5668. [Google Scholar] [CrossRef]
- Wen, P.; Huang, Y.; Qiu, Z.; Li, Q. Microbial response during treatment of different types of landfill leachate in a semi-aerobic aged refuse biofilter. Chemosphere 2021, 262, 127822. [Google Scholar] [CrossRef]
- Song, L.; Wang, Y.; Tang, W.; Lei, Y. Archaeal community diversity in municipal waste landfill sites. Appl. Microbiol. Biotechnol. 2015, 99, 6125–6137. [Google Scholar] [CrossRef]
- Gaspari, M.; Treu, L.; Zhu, X.; Palù, M.; Angelidaki, I.; Campanaro, S.; Kougias, P.G. Microbial dynamics in biogas digesters treating lipid-rich substrates via genome-centric metagenomics. Sci. Total Environ. 2021, 778, 146296. [Google Scholar] [CrossRef]
- Osman, J.R.; Wang, Y.; Jaubert, C.; Nguyen, T.N.; Fernandes, G.R.; DuBow, M.S. The bacterial communities of surface soils from desert sites in the eastern Utah (USA) portion of the Colorado Plateau. Microbiol. Res. 2021, 244, 126664. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ichino, T.; Saito, A. Transition of the Bacterial Community and Culturable Chitinolytic Bacteria in Chitin-treated Upland Soil: From Streptomyces to Methionine-auxotrophic Lysobacter and Other Genera. Microbes Environ. 2020, 35, ME19070. [Google Scholar] [CrossRef] [Green Version]
- Mwaikono, K.S.; Maina, S.; Sebastian, A.; Schilling, M.; Kapur, V.; Gwakisa, P. High-throughput sequencing of 16S rRNA Gene Reveals Substantial Bacterial Diversity on the Municipal Dumpsite. BMC Microbiol. 2016, 16, 145. [Google Scholar] [CrossRef] [Green Version]
- Christensen, P.; Cook, F.D. Lysobacter, a new genus of nonfruiting, gliding bacteria with a high base ratio. Int. J. Syst. Bacteriol. 1978, 28, 367–393. [Google Scholar] [CrossRef] [Green Version]
- Panthee, S.; Hamamoto, H.; Paudel, A.; Sekimizu, K. Lysobacter species: A potential source of novel antibiotics. Arch. Microbiol. 2016, 198, 839–845. [Google Scholar] [CrossRef]
- Horel, A.; Mortazavi, B.; Sobecky, P.A. Input of organic matter enhances degradation of weathered diesel fuel in sub-tropical sediments. Sci. Total Environ. 2015, 533, 82–90. [Google Scholar] [CrossRef]
- Pishgar, R.; Dominic, J.A.; Sheng, Z.; Tay, J.H. Denitrification performance and microbial versatility in response to different selection pressures. Bioresour. Technol. 2019, 281, 72–83. [Google Scholar] [CrossRef]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segers, P.; Vancanneyt, M.; Pot, B.; Torck, U.; Hoste, B.; Dewettinck, D.; Falsen, E.; Kersters, K.; De Vos, P. Classification of Pseudomonas diminuta Leifson and Hugh 1954 and Pseudomonas vesicularis Büsing, Döll, and Freytag 1953 in Brevundimonas gen. nov. as Brevundimonas diminuta comb. nov. and Brevundimonas vesicularis comb. nov., respectively. Int. J. Syst. Bacteriol. 1994, 44, 499–510. [Google Scholar] [CrossRef]
- Ryan, M.P.; Pembroke, J.T. Brevundimonas spp: Emerging global opportunistic pathogens. Virulence 2018, 9, 480–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, F.F.; Su, Y.; Wei, X.M.; He, Y.H.; Wu, Z.C.; Ghulam, A.; He, R. Diversity and activity of sulphur-oxidizing bacteria and sulphate-reducing bacteria in landfill cover soils. Lett. Appl. Microbiol. 2014, 59, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.; Park, J.; Lim, S.I.; Hur, H.G.; Kim, D.; Park, K. Size-resolved culturable airborne bacteria sampled in rice field, sanitary landfill, and waste incineration sites. J. Environ. Monit. 2010, 12, 1619–1624. [Google Scholar] [CrossRef]
- Amano, J.; Hase, R.; Otsuka, Y.; Tsuchimochi, T.; Noguchi, Y.; Igarashi, S. Catheter-related bloodstream infection by Microbacterium paraoxydans in a pediatric patient with B-cell precursor acute lymphocytic leukemia: A case report and review of literature on Microbacterium bacteremia. J. Infect. Chemother. 2019, 25, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Woo, G.K.; Yuen, K.Y. Catheter-related Microbacterium bacteremia identified by 16S rRNA gene sequencing. J. Clin. Microbiol. 2002, 40, 2681–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.C.; Kämpfer, P.; Chen, W.M.; Yen, W.S.; Arun, A.B.; Lai, W.A.; Shen, F.T.; Rekha, P.D.; Lin, K.Y.; Chou, J.H. Luteimonas composti sp. nov., a moderately thermophilic bacterium isolated from food waste. Int. J. Syst. Evol. Microbiol. 2007, 57, 741–744. [Google Scholar] [CrossRef]
- Rani, P.; Mukherjee, U.; Verma, H.; Kamra, K.; Lal, R. Luteimonas tolerans sp. nov., isolated from hexachlorocyclohexane-contaminated soil. Int. J. Syst. Evol. Microbiol. 2016, 66, 1851–1856. [Google Scholar] [CrossRef]
- Siddiqi, M.Z.; Yeon, J.M.; Choi, H.; Lee, J.H.; Kim, S.Y.; Wee, J.H.; Im, W.T. Luteimonas granuli sp. nov., Isolated from Granules of the Wastewater Treatment Plant. Curr. Microbiol. 2020, 77, 2002–2007. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Ma, J.; Xu, N.; Xin, F.; Zhang, W.; Zhang, H.; Dong, W.; Jiang, M. Luteimonas wenzhouensis Sp. Nov., A Chitinolytic Bacterium Isolated from a Landfill Soil. Curr. Microbiol. 2021, 78, 383–388. [Google Scholar] [CrossRef]
- Wang, X.; Cao, A.; Zhao, G.; Zhou, C.; Xu, R. Microbial community structure and diversity in a municipal solid waste landfill. Waste Manag. 2017, 66, 79–87. [Google Scholar] [CrossRef]
- Chen, B.; Yang, Y.; Liang, X.; Yu, K.; Zhang, T.; Li, X. Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef]
- Ben Maamar, S.; Glawe, A.J.; Brown, T.K.; Hellgeth, N.; Hu, J.; Wang, J.P.; Huttenhower, C.; Hartmann, E.M. Mobilizable antibiotic resistance genes are present in dust microbial communities. PLoS Pathog. 2020, 16, e1008211. [Google Scholar] [CrossRef]
- Pérez, J.; Contreras-Moreno, F.J.; Marcos-Torres, F.J.; Moraleda-Muñoz, A.; Muñoz-Dorado, J. The antibiotic crisis: How bacterial predators can help. Comput. Struct. Biotechnol. J. 2020, 18, 2547–2555. [Google Scholar] [CrossRef]
- Wu, D.; Huang, X.H.; Sun, J.Z.; Graham, D.W.; Xie, B. Antibiotic Resistance Genes and Associated Microbial Community Conditions in Aging Landfill Systems. Environ. Sci. Technol. 2017, 51, 12859–12861. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, W.; Qiao, J.; Song, L. Occurrence and prevalence of antibiotic resistance in landfill leachate. Environ. Sci. Pollut. Res. Int. 2015, 22, 12525–12533. [Google Scholar] [CrossRef]
- Zhao, R.; Feng, J.; Yin, X.; Liu, J.; Fu, W.; Berendonk, T.U.; Zhang, T.; Li, X.; Li, B. Antibiotic resistome in landfill leachate from different cities of China deciphered by metagenomic analysis. Water Res. 2018, 134, 126–139. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Li, Z.; Shen, D.; Feng, H.; Zhou, H.; Wang, M. Prevalence of fluoroquinolone, macrolide and sulfonamide-related resistance genes in landfills from East China, mainly driven by MGEs. Ecotoxicol. Environ. Saf. 2020, 190, 110131. [Google Scholar] [CrossRef]
- Kalwasińska, A.; Burkowska, A. Municipal landfill sites as sources of microorganisms potentially pathogenic to humans. Environ. Sci. Process. Impacts 2013, 15, 1078–1086. [Google Scholar] [CrossRef]
- De Mandal, S.; Mathipi, V.; Muthukumaran, R.B.; Gurusubramanian, G.; Lalnunmawii, E.; Kumar, N.S. Amplicon sequencing and imputed metagenomic analysis of waste soil and sediment microbiome reveals unique bacterial communities and their functional attributes. Environ. Monit. Assess. 2019, 191, 778. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhao, G.; Chao, X.; Xie, L.; Wang, H. The Characteristic of Virulence, Biofilm and Antibiotic Resistance of Klebsiella pneumoniae. Int. J. Environ. Res. Public Health 2020, 28, 6278. [Google Scholar] [CrossRef]
- Lee, C.R.; Lee, J.H.; Park, K.S.; Jeon, J.H.; Kim, Y.B.; Cha, C.J.; Jeong, B.C.; Lee, S.H. Antimicrobial Resistance of Hypervirulent Klebsiella pneumoniae: Epidemiology, Hypervirulence-Associated Determinants, and Resistance Mechanisms. Front. Cell Infect. Microbiol. 2017, 7, 483. [Google Scholar] [CrossRef] [Green Version]
- Trainor, E.A.; Nicholson, T.L.; Merkel, T.J. Bordetella pertussis transmission. Pathog. Dis. 2015, 73, ftv068. [Google Scholar] [CrossRef] [Green Version]
- Coote, J.G. Environmental sensing mechanisms in Bordetella. Adv. Microb. Physiol. 2001, 44, 141–181. [Google Scholar]
- Zhang, Y.; Ran, Z.; Tian, M.; Zhou, Y.; Yang, J.; Yin, J.; Lu, D.; Li, R.; Zhong, J. Commensal Microbes Affect Host Humoral Immunity to Bordetella pertussis Infection. Infect. Immun. 2019, 87, e00421-19. [Google Scholar] [CrossRef] [Green Version]
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Kuzi, S.; Blum, S.E.; Kahane, N.; Adler, A.; Hussein, O.; Segev, G.; Aroch, I. Multi-drug-resistant Acinetobacter calcoaceticus-Acinetobacter baumannii complex infection outbreak in dogs and cats in a veterinary hospital. J. Small Anim. Pract. 2016, 57, 617–625. [Google Scholar] [CrossRef]
- Nocera, F.P.; Attili, A.R.; De Martino, L. Acinetobacter baumannii: Its Clinical Significance in Human and Veterinary Medicine. Pathogens 2021, 10, 127. [Google Scholar] [CrossRef]
- Matereke, L.T.; Okoh, A.I. Listeria monocytogenes Virulence, Antimicrobial Resistance and Environmental Persistence: A Review. Pathogens 2020, 9, 528. [Google Scholar] [CrossRef] [PubMed]
- Lüth, S.; Halbedel, S.; Rosner, B.; Wilking, H.; Holzer, A.; Roedel, A.; Dieckmann, R.; Vincze, S.; Prager, R.; Flieger, A.; et al. Backtracking and forward checking of human listeriosis clusters identified a multiclonal outbreak linked to Listeria monocytogenes in meat products of a single producer. Emerg. Microbes Infect. 2020, 9, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 2010, 23, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloos, W.E.; Musselwhite, M.S. Distribution and persistence of Staphylococcus and Micrococcus species and other aerobic bacteria on human skin. Appl. Microbiol. 1975, 30, 381–385. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus epidermidis—The ‘accidental’ pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
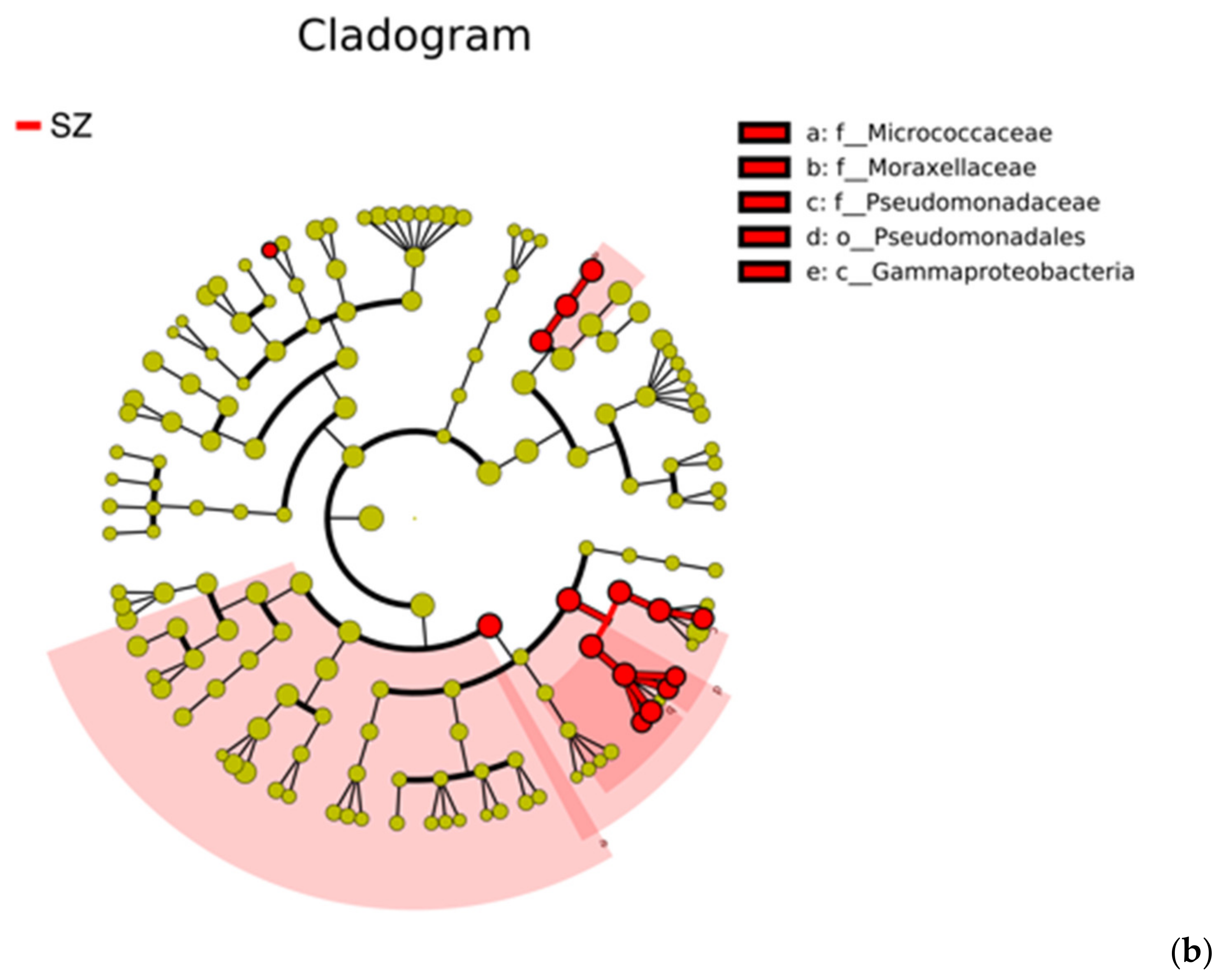{kind=link}
| Sample ID | Scaftigs | Total Length (bp) | Average Length (bp) | Max Length (bp) | N50 Length (bp) | GC% |
|---|---|---|---|---|---|---|
| SZ1 | 304,238 | 307,910,287 | 1012.07 | 73,348 | 1029 | 59.04 |
| SZ2 | 322,415 | 318,058,282 | 986.49 | 280,023 | 974 | 57.94 |
| SZ3 | 155,206 | 121,420,891 | 782.32 | 36,265 | 752 | 62.61 |
| WH1 | 333,139 | 398,161,179 | 1195.18 | 95,020 | 1355 | 54.52 |
| WH2 | 247,351 | 287,233,070 | 1161.24 | 179,900 | 1282 | 57.62 |
| WH3 | 188,206 | 227,855,957 | 1210.67 | 105,958 | 1339 | 51.01 |
| ARG Subtype | Drug Class | Microbial Taxa in Both Landfills | |
|---|---|---|---|
| SZ | WH | ||
| sul2 | Sulfonamide | Proteobacteria | Proteobacteria |
| tetX | Tetracycline | Proteobacteria | Deinococcus-Thermus; Actinobacteria |
| ErmF | MLS 1 | Unclassified | Bacteroidetes |
| sul1 | Sulfonamide | Unclassified | Bacteroidetes |
| OXA-347 | Multidrug | Unclassified | Unclassified |
| Erm35 | MLS | Bacteroidetes | Bacteroidetes |
| floR | Phenicol | Proteobacteria | Unclassified |
| aadA | Aminoglycoside | Unclassified | Unclassified |
| tet33 | Tetracycline | Actinobacteria | Actinobacteria |
| adeF | Multidrug | Proteobacteria | Proteobacteria |
| vanTmL | Glycopeptide | Proteobacteria; Bacteroidetes | Bacteroidetes |
| mphD | Macrolide | Bacteroidetes | Bacteroidetes |
| BcI | Multidrug | Not found | Proteobacteria |
| cmx | Phenicol | Unclassified | Unclassified |
| NDM-8 | Multidrug | Proteobacteria | Proteobacteria |
| APH2-IIIa | Aminoglycoside | Proteobacteria | Bacteroidetes |
| aadA3 | Aminoglycoside | Unclassified | Unclassified |
| blaR1 | Penam | Proteobacteria | Not found |
| APH3-Ib | Aminoglycoside | Proteobacteria | Proteobacteria |
| Erm42 | MLS | Proteobacteria | Proteobacteria |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, S.; Xia, M.; Tao, J.; Pang, Y.; Yu, F.; Wu, J.; Chen, S. Metagenomics Analysis Reveals the Microbial Communities, Antimicrobial Resistance Gene Diversity and Potential Pathogen Transmission Risk of Two Different Landfills in China. Diversity 2021, 13, 230. https://doi.org/10.3390/d13060230
Wan S, Xia M, Tao J, Pang Y, Yu F, Wu J, Chen S. Metagenomics Analysis Reveals the Microbial Communities, Antimicrobial Resistance Gene Diversity and Potential Pathogen Transmission Risk of Two Different Landfills in China. Diversity. 2021; 13(6):230. https://doi.org/10.3390/d13060230
Chicago/Turabian StyleWan, Shan, Min Xia, Jie Tao, Yanjun Pang, Fugen Yu, Jun Wu, and Shanping Chen. 2021. "Metagenomics Analysis Reveals the Microbial Communities, Antimicrobial Resistance Gene Diversity and Potential Pathogen Transmission Risk of Two Different Landfills in China" Diversity 13, no. 6: 230. https://doi.org/10.3390/d13060230