Increased Genetic Diversity via Gene Flow Provides Hope for Acacia whibleyana, an Endangered Wattle Facing Extinction

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
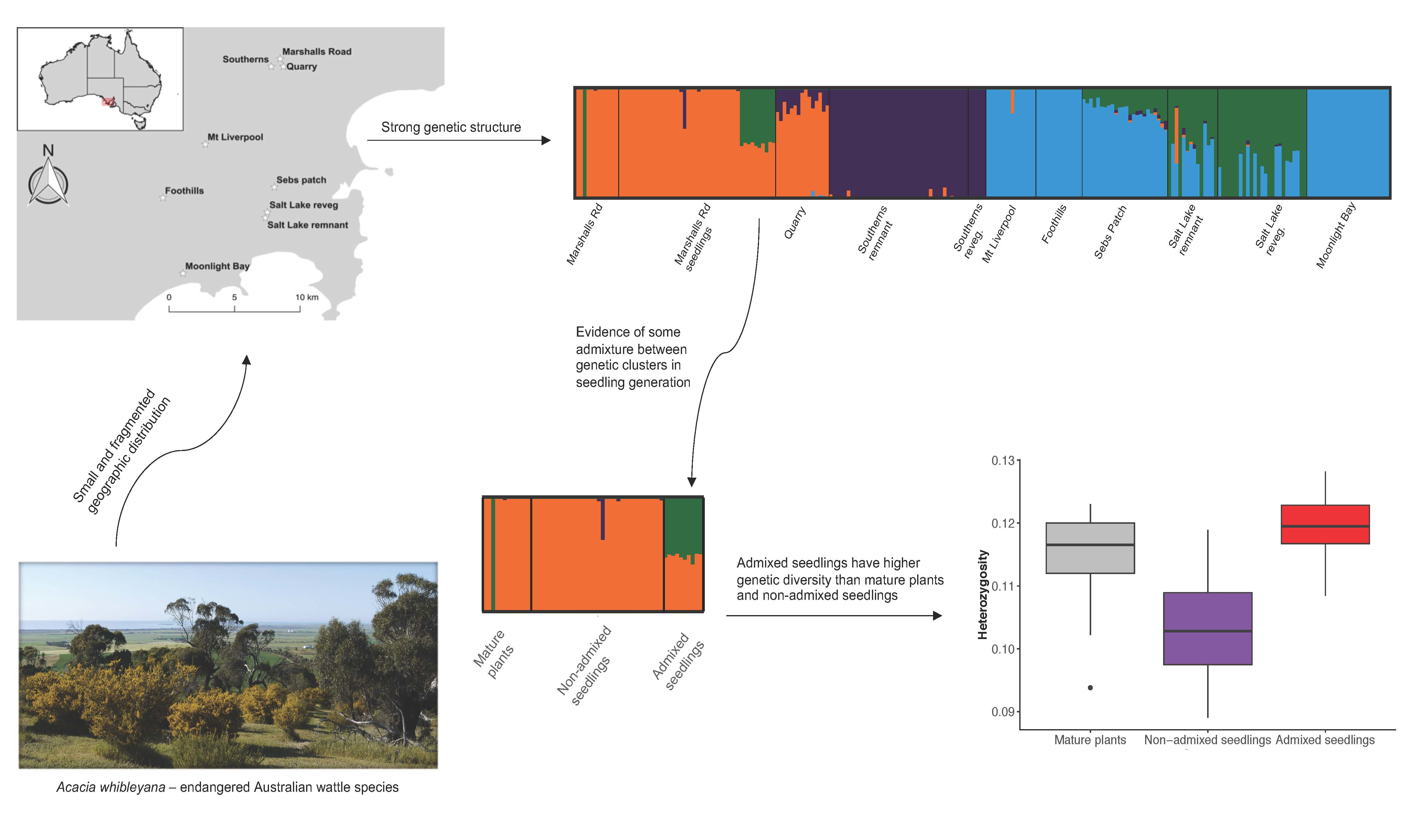
2.1. Study Species and Sampling
2.2. DNA Extraction, Sequencing and Filtering
2.3. SNP Calling
2.4. Genetic Structure Analysis
2.5. Genetic Diversity and Inbreeding Analysis
2.6. Redundancy Analysis
3. Results
3.1. Genetic Structure Analysis
3.2. Genetic Diversity and Inbreeding
4. Discussion
4.1. Genetic Structure and Sources of Genetic Variation
4.2. Opportunistic Test of Admixture on Genetic Diversity
4.3. Genetic Diversity and Inbreeding
4.4. Management Recommendations
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Stand | Habitat Type | Census Count | n | Land Use History |
|---|---|---|---|---|
| Marshalls Road | Roadside vegetation. | 42 reveg. | 12 | Road reserve. Control burn in 2018 resulted in A. whibleyana plants emerging post fire. |
| 100+ seedlings | 44 | |||
| Quarry | Post-disturbance regeneration in disused quarry, surrounded by remnant eucalypt woodland | 189 remnant | 15 | Council-owned land. Quarried up to 1950’s, stockpiling gravel then woodchips (to 1993). |
| Southerns | Remnant eucalypt woodland. | 59 remnant | 39 | Private land. 40ha fenced to exclude stock in May 2012. |
| 8 reveg. | 5 | |||
| Mount Liverpool | Remnant eucalypt woodland. | 50 remnant | 14 | Private land subdivided for rural living & stock removed 2005. |
| Foothills Road | Roadside vegetation. | 17 remnant | 13 | Road reserve, burnt in 2005 bushfires. A. whibleyana plants emerged post fire. |
| Sebs Patch | Remnant chenopod scrub along salt-scalded drainage line and salt lake. | 107 remnant | 24 | Private land under different land management regimes (grazed by sheep intermittently, a small portion fenced & revegetated) |
| Salt Lake | Remnant chenopod scrub adjacent to salt lake. | 137 remnant | 14 | Council-owned land of road intersection, fenced in 1995 to exclude vehicles and rabbits. |
| 26 reveg. | 25 | |||
| Moonlight Bay | Remnant mallee woodland on salt lake lunettes. | 557 remnant | 23 | Private land. Approx. one third of population fenced from stock. Remainder accessible to sheep on intermittent basis. Area around salt lake burnt in 2005 bushfires. |


References
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Nicotra, A.B.; Beever, E.A.; Robertson, A.L.; Hofmann, G.E.; O’Leary, J. Assessing the components of adaptive capacity to improve conservation and management efforts under global change. Conserv. Biol. 2015, 29, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Leimu, R.; Mutikainen, P.; Koricheva, J.; Fischer, M. How general are positive relationships between plant population size, fitness and genetic variation? J. Ecol. 2006, 94, 942–952. [Google Scholar] [CrossRef]
- Aguilar, R.; Quesada, M.; Ashworth, L.; Herrerías-Diego, Y.; Lobo, J. Genetic consequences of habitat fragmentation in plant populations: Susceptible signals in plant traits and methodological approaches. Mol. Ecol. 2008, 17, 5177–5188. [Google Scholar] [CrossRef]
- Keller, L. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [Green Version]
- Weeks, A.; Sgrò, C.M.; Young, A.G.; Frankham, R.; Mitchell, N.J.; Miller, K.A.; Byrne, M.; Coates, D.J.; Eldridge, M.D.B.; Sunnucks, P.; et al. Assessing the benefits and risks of translocations in changing environments: A genetic perspective. Evol. Appl. 2011, 4, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Barmentlo, H.; Meirmans, P.G.; Luijten, S.H.; Triest, L.; Oostermeijer, G. Outbreeding depression and breeding system evolution in small, remnant populations of Primula vulgaris: Consequences for genetic rescue. Conserv. Genet. 2017, 19, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.B.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the probability of outbreeding depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef]
- Ralls, K.; Dudash, M.R.; Eldridge, M.D.B.; Fenster, C.B.; Sunnucks, P.; Ballou, J.D.; Lacy, R.; Frankham, R. Call for a paradigm shift in the genetic management of fragmented populations. Conserv. Lett. 2017, 11, e12412. [Google Scholar] [CrossRef]
- Pickup, M.; Field, D.L.; Rowell, D.M.; Young, A.G. Source population characteristics affect heterosis following genetic rescue of fragmented plant populations. Proc. R. Soc. B Biol. Sci. 2013, 280, 20122058. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Sgrò, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A.; Robinson, Z.L.; Funk, W.C.; Fitzpatrick, S.W.; Allendorf, F.W.; Tallmon, D.A.; Whiteley, A.R. The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol. Evol. 2019, 34, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.V.; Nevill, P.G.; Krauss, S.L. Next generation restoration genetics: Applications and opportunities. Trends Plant Sci. 2014, 19, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, Y.G.S.; Vitorino, L.C.; De Souza, U.J.B.; Bessa, L.A. Recent trends in research on the genetic diversity of plants: Implications for conservation. Diversity 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Breed, M.F.; Harrison, P.A.; Blyth, C.; Byrne, M.; Gaget, V.; Gellie, N.J.C.; Groom, S.V.; Hodgson, R.; Mills, J.G.; Prowse, T.A.A.; et al. The potential of genomics for restoring ecosystems and biodiversity. Nat. Rev. Genet. 2019, 20, 615–628. [Google Scholar] [CrossRef]
- Ottewell, K.; Bickerton, D.C.; Byrne, M.; Lowe, A.J. Bridging the gap: A genetic assessment framework for population-level threatened plant conservation prioritization and decision-making. Divers. Distrib. 2015, 22, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Commonwealth of Australia. Threatened Species Strategy—Year One Report; Department of the Environment and Energy: Alice Springs, NT, Australia, 2016.
- Jusaitis, M. Translocation trials confirm specific factors affecting the establishment of three endangered plant species. Ecol. Manag. Restor. 2005, 6, 61–67. [Google Scholar] [CrossRef]
- Jusaitis, M.; Polomka, L. Weeds and propagule type influence translocation success in the endangered Whibley wattle, Acacia whibleyana (Leguminosae: Mimosoideae). Ecol. Manag. Restor. 2008, 9, 72–76. [Google Scholar] [CrossRef]
- Faast, R.; Blyth, C.; Breed, M.F.; Packer, J.G. Acacia Whibleyana: Part 1—Literature Synthesis to Inform Conservation of a Threatend Acacia; Eyre Peninsula Natural Resources Management Board: Port Lincoln, South Australia, 2019.
- Adams, M.; Attiwill, P. Role of Acacia spp. in nutrient balance and cycling in regenerating Eucalyptus regnans F. Muell. forests. I. Temporal changes in biomass and nutrient content. Aust. J. Bot. 1984, 32, 205–215. [Google Scholar] [CrossRef]
- Jusaitis, M.; Sorensen, B. Conservation Biology of Acacia Whibleyana; Black Hill Flora Centre, Botanic Gardens of Adelaide: Adelaide, Australia, 1998.
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.A.; Postlethwait, J.H. Stacks: Building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.M.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.B.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Meirmans, P.G. genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 2020. [Google Scholar] [CrossRef] [Green Version]
- Shafer, A.B.A.; Peart, C.R.; Tusso, S.; Maayan, I.; Brelsford, A.; Wheat, C.W.; Wolf, J.B.W. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol. 2016, 8, 907–917. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, V.B. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Ottewell, K.; Bickerton, D.; Lowe, A.J. Can a seed bank provide demographic and genetic rescue in a declining population of the endangered shrub Acacia pinguifolia? Conserv. Genet. 2010, 12, 669–678. [Google Scholar] [CrossRef]
- Stone, G.N.; Raine, N.E.; Prescott, M.; Willmer, P.G. Pollination ecology of acacias (Fabaceae, Mimosoideae). Aust. Syst. Bot. 2003, 16, 103–118. [Google Scholar] [CrossRef]
- Breed, M.F.; Ottewell, K.; Gardner, M.G.; Marklund, M.H.K.; E Dormontt, E.; Lowe, A.J. Mating patterns and pollinator mobility are critical traits in forest fragmentation genetics. Heredity 2013, 115, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Jusaitis, M.; Sorensen, B.; Polomka, L. Population structure, flowering and seed production in the endangered whibley wattle, Acacia whibleyana (Leguminosae: Mimosoideae). Open Conserv. Biol. J. 2009, 3, 4–13. [Google Scholar] [CrossRef]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2004, 14, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Borrell, J.S.; Wang, N.; Nichols, R.A.; Buggs, R. Genetic diversity maintained among fragmented populations of a tree undergoing range contraction. Heredity 2018, 121, 304–318. [Google Scholar] [CrossRef] [Green Version]
- De Kort, H.; Vandepitte, K.; Bruun, H.H.; Closset-Kopp, D.; Honnay, O.; Mergeay, J. Landscape genomics and a common garden trial reveal adaptive differentiation to temperature across Europe in the tree speciesAlnus glutinosa. Mol. Ecol. 2014, 23, 4709–4721. [Google Scholar] [CrossRef]
- Johnson, J.; Gaddis, K.D.; Cairns, D.M.; Konganti, K.; Krutovsky, K.V. Landscape genomic insights into the historic migration of mountain hemlock in response to Holocene climate change. Am. J. Bot. 2017, 104, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Pais, A.; Whetten, R.W.; Xiang, Q. (Jenny) Ecological genomics of local adaptation in Cornus florida L. by genotyping by sequencing. Ecol. Evol. 2016, 7, 441–465. [Google Scholar] [CrossRef] [PubMed]
- He, T.; D’Agui, H.; Lim, S.L.; Enright, N.J.; Luo, Y. Evolutionary potential and adaptation of Banksia attenuata (Proteaceae) to climate and fire regime in southwestern Australia, a global biodiversity hotspot. Sci. Rep. 2016, 6, 26315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenrick, J.; Knox, R.B. Quantitative analysis of self-incompatibility in trees of seven species of Acacia. J. Hered. 1989, 80, 240–245. [Google Scholar] [CrossRef]
- Kenrick, J.; Kaul, V.; Williams, E.G. Self-incompatibility in Acacia retinodes: Site of pollen-tube arrest is the nucellus. Planta 1986, 169, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Millar, M.A.; Byrne, M.; Nuberg, I.; Sedgley, M. High outcrossing and random pollen dispersal in a planted stand of Acacia saligna subsp. saligna revealed by paternity analysis using microsatellites. Tree Genet. Genomes 2007, 4, 367–377. [Google Scholar] [CrossRef]
- Lesica, P.; Allendorf, F.W. When are peripheral populations valuable for conservation? Conserv. Biol. 1995, 9, 753–760. [Google Scholar] [CrossRef]
- Coates, D.J.; Tischler, G.; McComb, J.A. Genetic variation and the mating system in the rare Acacia sciophanes compared with its common sister species Acacia anfractuosa (Mimosaceae). Conserv. Genet. 2006, 7, 931–944. [Google Scholar] [CrossRef]
- Nano, C.; Nano, T.; Gibson, J.; Pavey, C. Recovery Action Implementation for Threatened Arid Acacias: Distribution, Monitoring and Indigenous Ecological Knowledge of A. Peuce, A. Undoolyana, A. Pickardii and A. Latzii; Department of Natural Resources, Environment, Arts & Sport Northern Territory Government: Alice Springs, Australia, 2008.


| Collection Stand | Marshalls Rd | Marshalls Rd Seedlings | Quarry | Southerns Remnant | Southerns reveg. | Mt Liverpool | Foothills | Sebs Patch | Salt Lake remnant | Salt Lake reveg. | Moonlight Bay |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Marshalls Rd | 0 | ||||||||||
| Marshalls Rd seedlings | −0.008 | 0 | |||||||||
| Quarry | 0.017 | 0.032 | 0 | ||||||||
| Southerns remnant | 0.090 | 0.101 | 0.087 | 0 | |||||||
| Southerns reveg. | 0.101 | 0.114 | 0.101 | 0.013 | 0 | ||||||
| Mt Liverpool | 0.156 | 0.158 | 0.153 | 0.122 | 0.145 | 0 | |||||
| Foothills | 0.192 | 0.193 | 0.189 | 0.147 | 0.176 | 0.185 | 0 | ||||
| Sebs Patch | 0.109 | 0.117 | 0.111 | 0.083 | 0.090 | 0.104 | 0.134 | 0 | |||
| Salt Lake remnant | 0.088 | 0.099 | 0.097 | 0.073 | 0.077 | 0.100 | 0.129 | 0.037 | 0 | ||
| Salt Lake reveg. | 0.115 | 0.123 | 0.126 | 0.097 | 0.104 | 0.122 | 0.148 | 0.061 | 0.000 | 0 | |
| Moonlight Bay | 0.136 | 0.145 | 0.137 | 0.111 | 0.116 | 0.135 | 0.161 | 0.084 | 0.075 | 0.097 | 0 |
| Source of Variation | Nested in | %var | F-Value | SD | c.i.95% |
|---|---|---|---|---|---|
| Within Individual | - | 73.6 | 0.263 | 0.002 | ±0.004 |
| Among Individual | Collection stand | 15.7 | 0.175 | 0.002 | ±0.004 |
| Among Population | 10.7 | 0.107 | 0.001 | ±0.002 |
| Collection Stand | Status | n | HO | HE | F | Kinship |
|---|---|---|---|---|---|---|
| All stands | ||||||
| Marshalls Rd | Translocation | 12 | 0.114 (0.111 to 0.116) b,c | 0.137 (0.135 to 0.140) c,d | 0.173 (0.163 to 0.182) b | −0.008 (0.018 to −0.027) a,b |
| Marshalls Rd seedlings (all) | Seedlings | 44 | 0.107 (0.105 to 0.109) a,b | 0.131 (0.128 to 0.133) b | 0.183 (0.176 to 0.189) b | 0.001 (0.003 to −0.002) a |
| Quarry | Remnant | 15 | 0.111 (0.109 to 0.114) b | 0.136 (0.134 to 0.139) c,d | 0.183 (0.174 to 0.192) b | 0.004 (0.011 to −0.003) a |
| Southerns remnant | Remnant | 39 | 0.118 (0.116 to 0.121) c | 0.145 (0.142 to 0.147) d | 0.183 (0.177 to 0.189) b | 0.000 (0.003 to −0.003) a |
| Southerns reveg. | Revegetation | 5 | 0.118 (0.115 to 0.121) c | 0.140 (0.137 to 0.143) c,d | 0.157 (0.143 to 0.170) b | 0.006 (0.043 to −0.031) a,b |
| Mt Liverpool | Remnant | 14 | 0.109 (0.107 to 0.112) b | 0.123 (0.120 to 0.126) a | 0.110 (0.100 to 0.121) a | 0.009 (0.026 to −0.007) a |
| Foothills | Remnant | 13 | 0.102 (0.099 to 0.105) a | 0.117 (0.114 to 0.120) a | 0.130 (0.119 to 0.141) a | 0.008 (0.023 to −0.006) a |
| Sebs Patch | Remnant | 24 | 0.109 (0.107 to 0.111) b | 0.138 (0.136 to 0.141) c,d | 0.211 (0.204 to 0.218) c | 0.001 (0.006 to −0.003) a |
| Salt Lake remnant | Remnant | 14 | 0.110 (0.108 to 0.112) b | 0.141 (0.139 to 0.144) d | 0.221 (0.212 to 0.229) c | −0.011 (−0.008 to −0.014) b |
| Salt Lake reveg. | Revegetation | 25 | 0.117 (0.115 to 0.120) b,c | 0.138 (0.136 to 0.141) c,d | 0.151 (0.144 to 0.159) b | 0.001 (0.009 to −0.006) a |
| Moonlight Bay | Remnant | 23 | 0.104 (0.102 to 0.106) a | 0.135 (0.132 to 0.137) b,c | 0.226 (0.219 to 0.234) c | −0.004 (0.000 to −0.008) a,b |
| Marshalls Rd focus | ||||||
| Marshalls Rd adults | Translocation | 12 | 0.114 (0.111 to 0.116) b | 0.137 (0.135 to 0.140) b | 0.173 (0.163 to 0.182) b | −0.008 (0.018 to −0.027) a |
| Marshalls Rd seedlings (non-admixed only) | Seedlings | 34 | 0.103 (0.101 to 0.106) a | 0.126 (0.124 to 0.129) a | 0.181 (0.173 to 0.188) b | 0.008 (0.010 to −0.002) a |
| Marshalls Rd seedlings (admixed only) | Seedlings | 10 | 0.120 (0.117 to 0.123) c | 0.125 (0.122 to 0.127) a | 0.036 (0.023 to 0.049) a | 0.117 (0.122 to −0.005) a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blyth, C.; Christmas, M.J.; Bickerton, D.C.; Faast, R.; Packer, J.G.; Lowe, A.J.; Breed, M.F. Increased Genetic Diversity via Gene Flow Provides Hope for Acacia whibleyana, an Endangered Wattle Facing Extinction. Diversity 2020, 12, 299. https://doi.org/10.3390/d12080299
Blyth C, Christmas MJ, Bickerton DC, Faast R, Packer JG, Lowe AJ, Breed MF. Increased Genetic Diversity via Gene Flow Provides Hope for Acacia whibleyana, an Endangered Wattle Facing Extinction. Diversity. 2020; 12(8):299. https://doi.org/10.3390/d12080299
Chicago/Turabian StyleBlyth, Colette, Matthew J. Christmas, Doug C. Bickerton, Renate Faast, Jasmin G. Packer, Andrew J. Lowe, and Martin F. Breed. 2020. "Increased Genetic Diversity via Gene Flow Provides Hope for Acacia whibleyana, an Endangered Wattle Facing Extinction" Diversity 12, no. 8: 299. https://doi.org/10.3390/d12080299