
Structural Characterization of 4-(4-Nitrophenyl)thiomorpholine, a Precursor in Medicinal Chemistry

Abstract
:1. Introduction
2. Results and Discussion
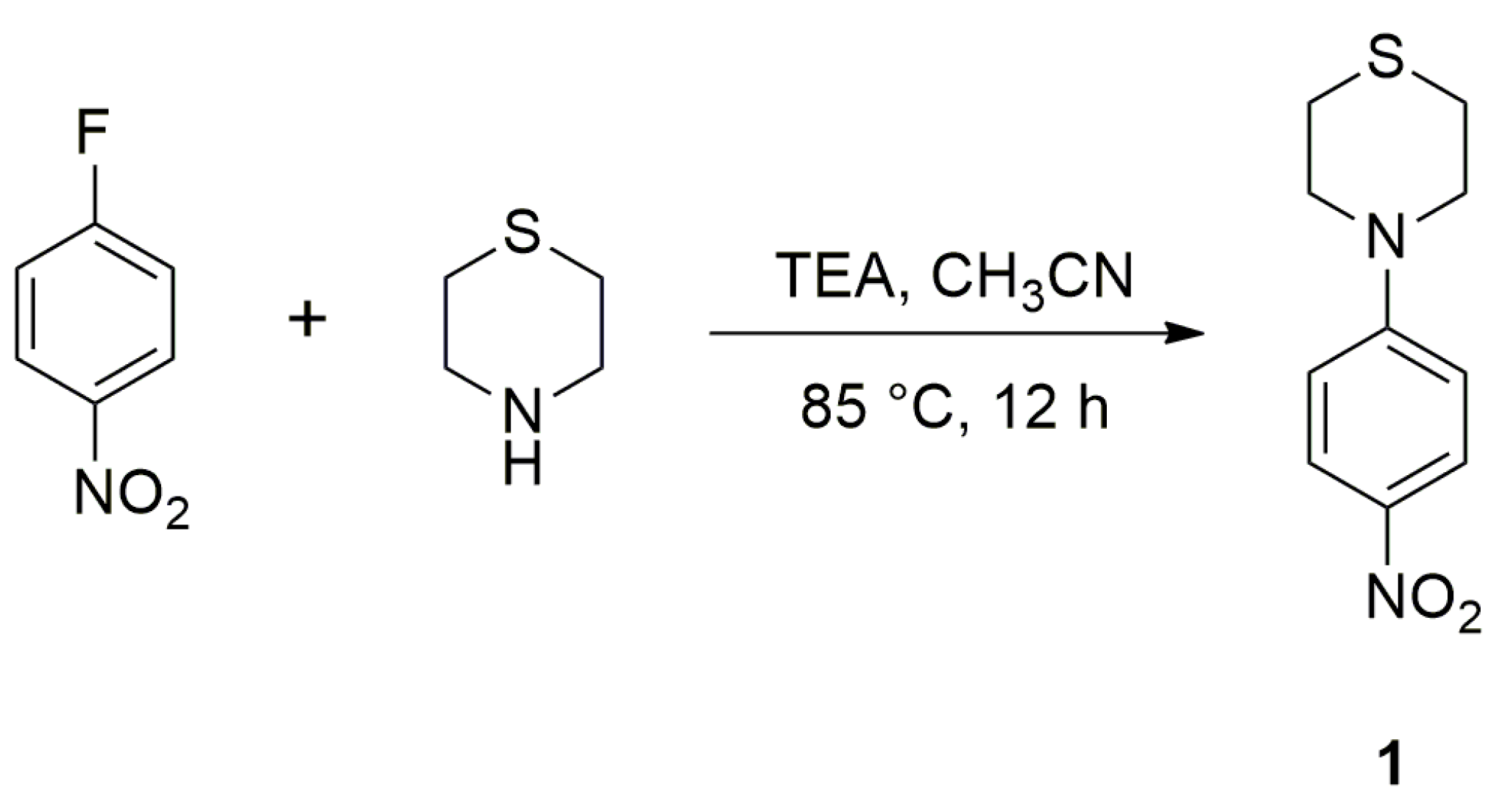
2.1. Synthesis
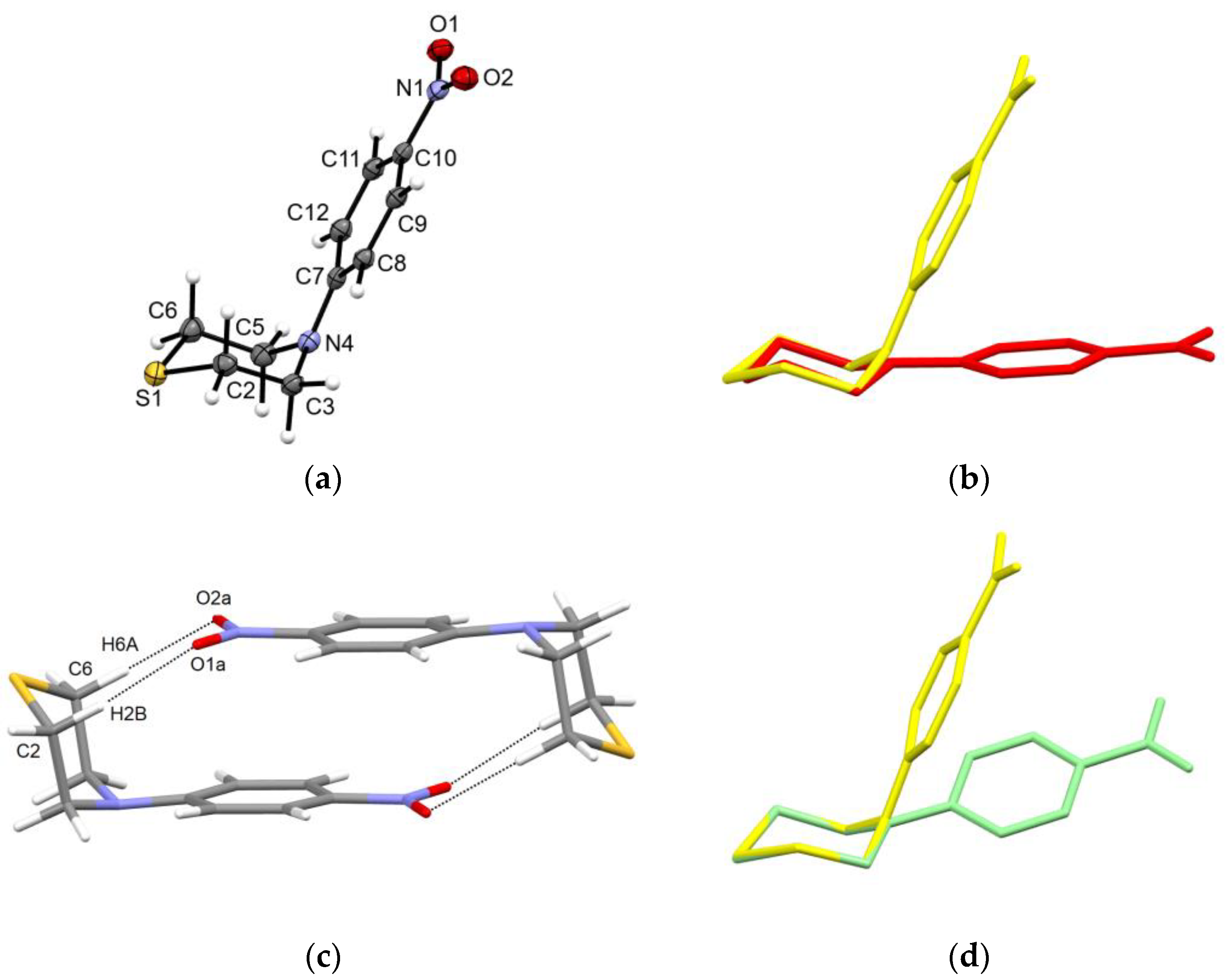
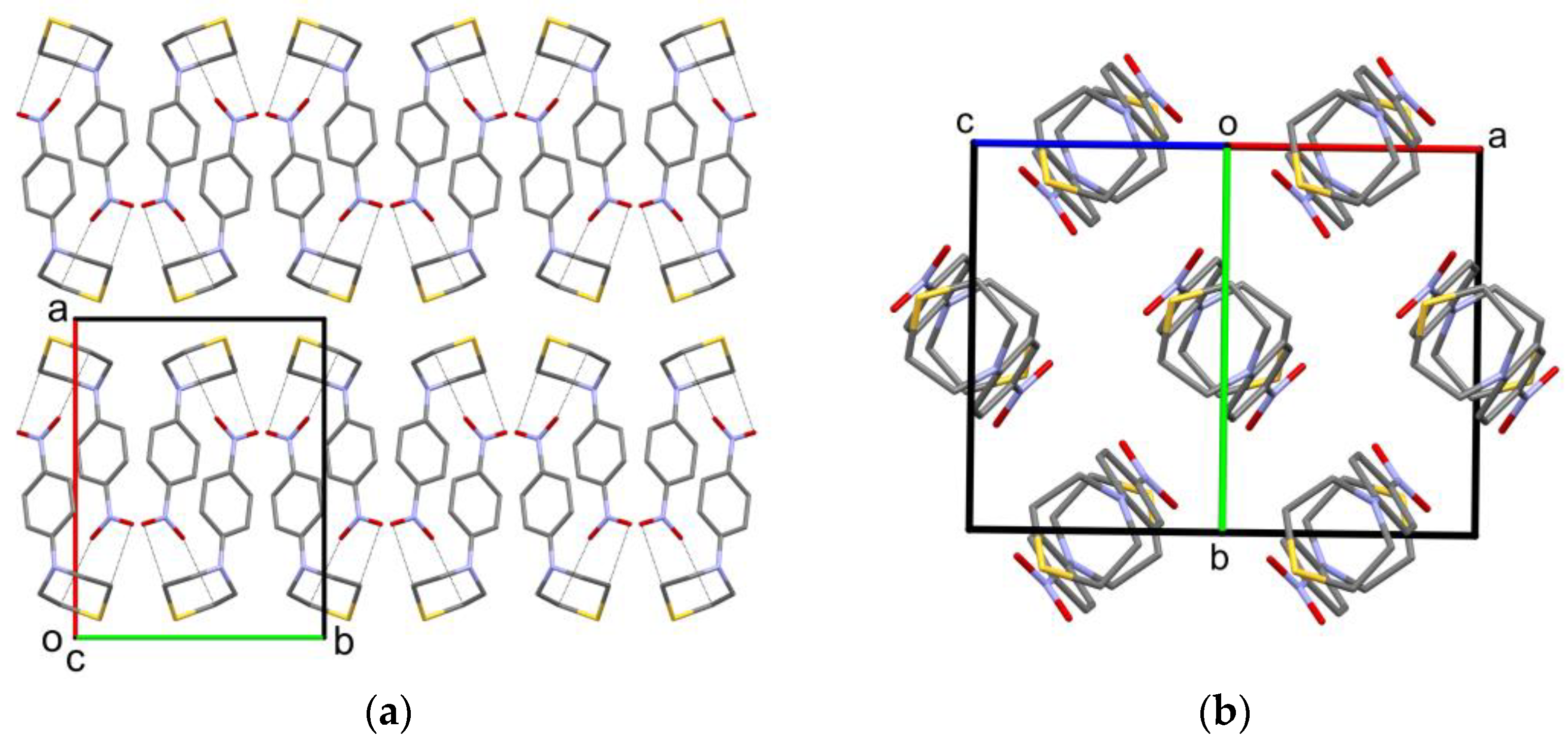
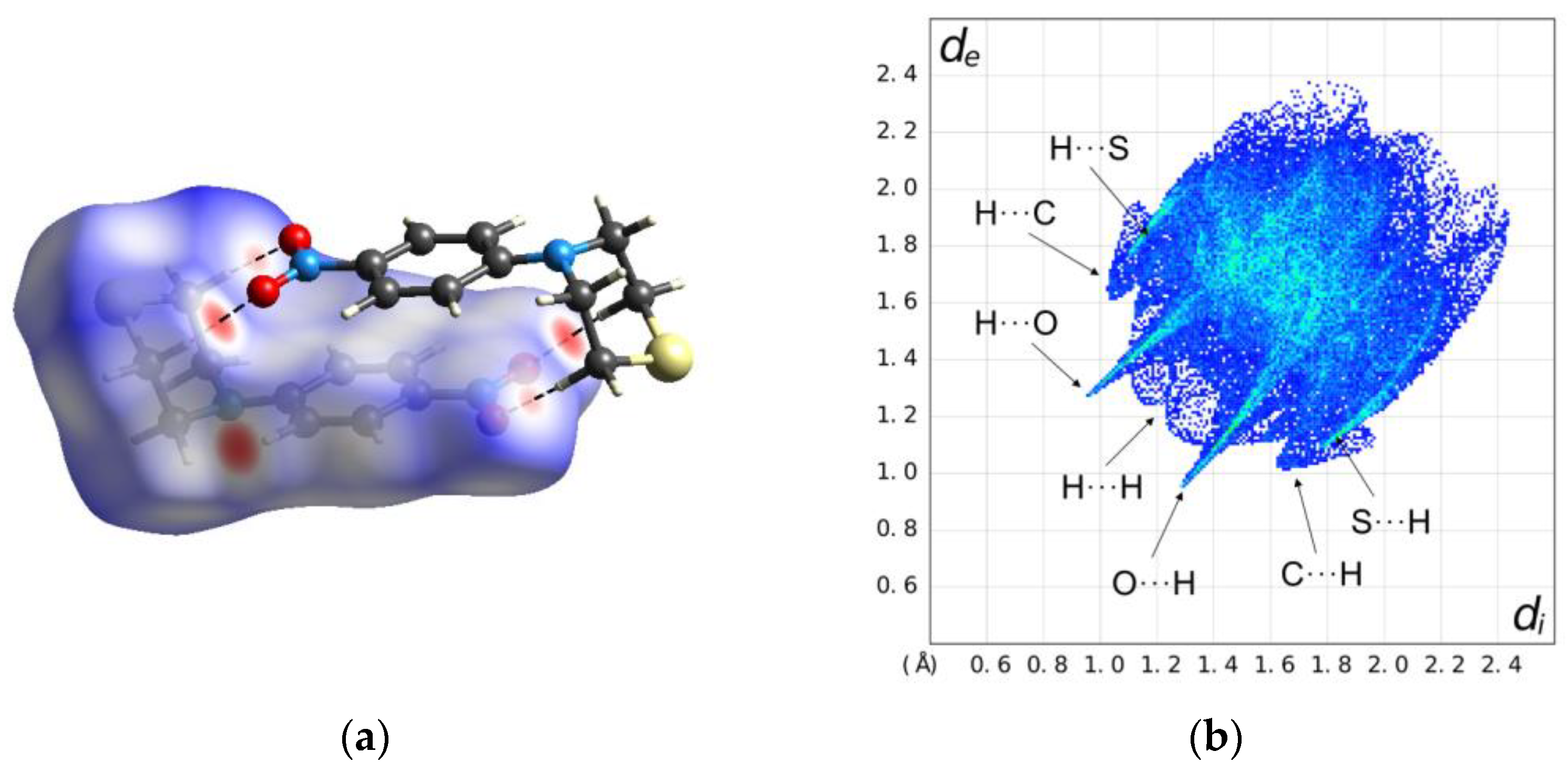
2.2. Solid-State Structure
3. Materials and Methods
3.1. General
3.2. Synthesis
3.3. X-ray Crystallography
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwink, L.; Stengelin, S.; Gossel, M. WO2003015769; Preparation of Indol-5-ylureas and Relate Compounds for the Treatment of Obesity and Type II Diabetes. 2003.
- Chapdelaine, M.; Davenport, T.; Haeberlein, M.; Horchler, C.; McCauley, J.; Pierson, E.; Sohn, D. WO2002055014; Preparation of Piperazinylchromans as 5-HT1B and 5-HT1D Agonists/Antagonists Useful as Antimigraine Drugs. 2002.
- Li, Y.; Ye, T.; Xu, L.; Dong, Y.; Luo, Y.; Wang, C.; Han, Y.; Chen, K.; Qin, M.; Liu, Y.; et al. Discovery of 4-piperazinyl-2-aminopyrimidine derivatives as dual inhibitors of JAK2 and FLT3. Eur. J. Med. Chem. 2019, 181, 111590. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wang, C.; Xin, X.; Li, S.; Li, Z.; Zhao, Y.; Gong, P. Design, synthesis and biological evaluation of novel 2,4-diaminopyrimidine derivatives as potent antitumor agents. New J. Chem. 2019, 43, 10190–10202. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, L.; Li, Z.; Shen, Q.; Xu, Q.; Li, W.; Liu, Y.; Gong, P. Design, synthesis and biological evaluation of novel 7-amino-[1,2,4]triazolo[4,3-f]pteridinone, and 7-aminotetrazolo[1,5-f]pteridinone derivative as potent antitumor agents. Eur. J. Med. Chem. 2019, 163, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.W.; Chan, J.H.; Freeman, G.A.; Romines, K.R.; Tidwell, J.H. Preparation of benzophenones and phenyl heteroaryl ketones as inhibitors of reverse transcriptase 2001.
- Gordeev, M.F.; Renslo, A.; Luehr, G.W.; Lam, S.; Westlund, N.E.; Patel, D.V. Preparation of Antibacterial Dihydrothiazinyl and Dihydrothiopyranyl Oxazolidinones 2003.
- Thomson, S.P.; Davies, R.T.; Allanson, N.M.; Kuvshinov, A.; Davies, G.M.; Edwards, P.N. WO2006123145; Preparation of Indolizinyloxoacetamides and Related Compounds as Medical and Agrochemical Fungicides. 2006.
- Downham, R.; Sibley, G.E.M.; Payne, L.J.; Edwards, P.; Davies, G.M. WO2008062182; 2-[(2-Substituted)-Indolizolin-3-yl]-2-Oxoacetamide Derivatives as Antifungal Agents and Their Preparation, Pharmaceutical and Agricultural Compositions and Use in the Treatment of Fungal Diseases. 2008.
- Birch, M.; Sibley, G.E.M.; Law, D.; Oliver, J.D. WO2009144473; Preparation of Indolizine Compounds for Use in Antifungal Combination Therapy. 2009.
- Mai, A.; Sbardella, G.; Artico, M.; Massa, S.; Lampis, G.; Deidda, D.; Pompei, R. N-[4-(1,1′-biphenylyl)methyl]-4-(4-thiomorpholinylmethyl) benzenamines, a new class of synthetic antituberculosis agents active against Mycobacterium avium. Med. Chem. Res. 1999, 9, 149–161. [Google Scholar]
- Courbon, G.M.; Palme, P.R.; Mann, L.; Richter, A.; Imming, P.; Rubinstein, J.L. Mechanism of mycobacterial ATP synthase inhibition by squaramides and second generation diarylquinolines. EMBO J. 2023, 42, e113687. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Trump, R.P.; Blanc, J.-B.E.; Stewart, E.L.; Brown, P.J.; Caivano, M.; Gray, D.W.; Hoekstra, W.J.; Willson, T.M.; Han, B.; Turnbull, P. Design and Synthesis of an Array of Selective Androgen Receptor Modulators. J. Comb. Chem. 2007, 9, 107–114. [Google Scholar] [CrossRef]
- Beach, S.F.; Hepworth, J.D.; Sawyer, J.; Hallas, G.; Marsden, R.; Mitchell, M.M.; Ibbitson, D.A.; Jones, A.M.; Neal, G.T. Dipole moments of some N-phenyl-substituted derivatives of pyrrolidine, piperidine, morpholine, and thiomorpholine. J. Chem.Soc. Perkin Trans. 2 1984, 217–221. [Google Scholar] [CrossRef]
- Purkait, N.; Kervefors, G.; Linde, E.; Olofsson, B. Regiospecific N-Arylation of Aliphatic Amines under Mild and Metal-Free Reaction Conditions. Angew. Chem. Int. Ed. Engl. 2018, 57, 11427–11431. [Google Scholar] [CrossRef]
- White, J.M.; Fellows, T. CSD Communication 2016. [CrossRef]
- White, J.M.; Skene, C.E.; Deadman, J.; Epa, R.; Foenander, S.; Hamer, K.; Fellowes, T.; Lim, S.F.; Marcuccio, S.M.; Martin, R.F. Synthesis and Structural Investigation of Some Electron-Rich Nitroaromatics. Aust. J. Chem. 2019, 72, 311–327. [Google Scholar] [CrossRef]
- Wang, L.J.; Li, W.W.; Yang, S.Y.; Yang, L. 4-(4-Nitro-phen-yl)morpholine. Acta Crystallogr. E 2012, 68, o1235. [Google Scholar] [CrossRef]
- Thakuria, R.; Sarma, B.; Nangia, A. 7.03-Hydrogen Bonding in Molecular Crystals. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Ed.; Elsevier: Oxford, UK, 2017; pp. 25–48. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Kitajgorodskij, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Bruker. APEX4; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- Bruker. SAINT V8.40B; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Bruker. SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Midgley, L.; Bourhis, L.J.; Dolomanov, O.V.; Grabowsky, S.; Kleemiss, F.; Puschmann, H.; Peyerimhoff, N. Vanishing of the atomic form factor derivatives in non-spherical structural refinement—A key approximation scrutinized in the case of Hirshfeld atom refinement. Acta Crystallogr. A 2021, 77, 519–533. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Crystallogr. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-ray | DFT | |
|---|---|---|
| C2–C3 | 1.5249(18) | 1.527 |
| C2–S1 | 1.8193(13) | 1.818 |
| C3–N4 | 1.4681(16) | 1.459 |
| C5–C6 | 1.5271(18) | 1.523 |
| C5–N4 | 1.4632(16) | 1.466 |
| C6–S1 | 1.8178(15) | 1.819 |
| C7–N4 | 1.3804(15) | 1.390 |
| S1–C2–C3 | 111.06(9) | 113.53 |
| N4–C3–C2 | 111.58(10) | 111.93 |
| N4–C5–C6 | 111.24(11) | 112.93 |
| S1–C6–C5 | 111.60(9) | 111.96 |
| C5–N4–C3 | 111.49(10) | 114.28 |
| C6–S1–C2 | 99.56(6) | 96.49 |
| C3–N4–C7–C8 | 14.86(12) | 10.93 |
| C5–N4–C7–C12 | –21.08(13) | 41.71 |
| D–H···A | d(D–H) | d(H–A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| C2–H2B···O1a | 1.100(16) | 2.313(16) | 3.4086(15) | 173.4(12) |
| C6–H6A···O2a | 1.072(18) | 2.498(18) | 3.5126(16) | 157.5(14) |
| C8–H8···O1b | 1.075(15) | 2.249(15) | 3.3023(15) | 165.9(12) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palme, P.R.; Goddard, R.; Imming, P.; Seidel, R.W. Structural Characterization of 4-(4-Nitrophenyl)thiomorpholine, a Precursor in Medicinal Chemistry. Molbank 2024, 2024, M1795. https://doi.org/10.3390/M1795
Palme PR, Goddard R, Imming P, Seidel RW. Structural Characterization of 4-(4-Nitrophenyl)thiomorpholine, a Precursor in Medicinal Chemistry. Molbank. 2024; 2024(1):M1795. https://doi.org/10.3390/M1795
Chicago/Turabian StylePalme, Paul R., Richard Goddard, Peter Imming, and Rüdiger W. Seidel. 2024. "Structural Characterization of 4-(4-Nitrophenyl)thiomorpholine, a Precursor in Medicinal Chemistry" Molbank 2024, no. 1: M1795. https://doi.org/10.3390/M1795