
2-(N-allylsulfamoyl)-N-propylbenzamide

Abstract
:1. Introduction
2. Results
2.1. Synthesis
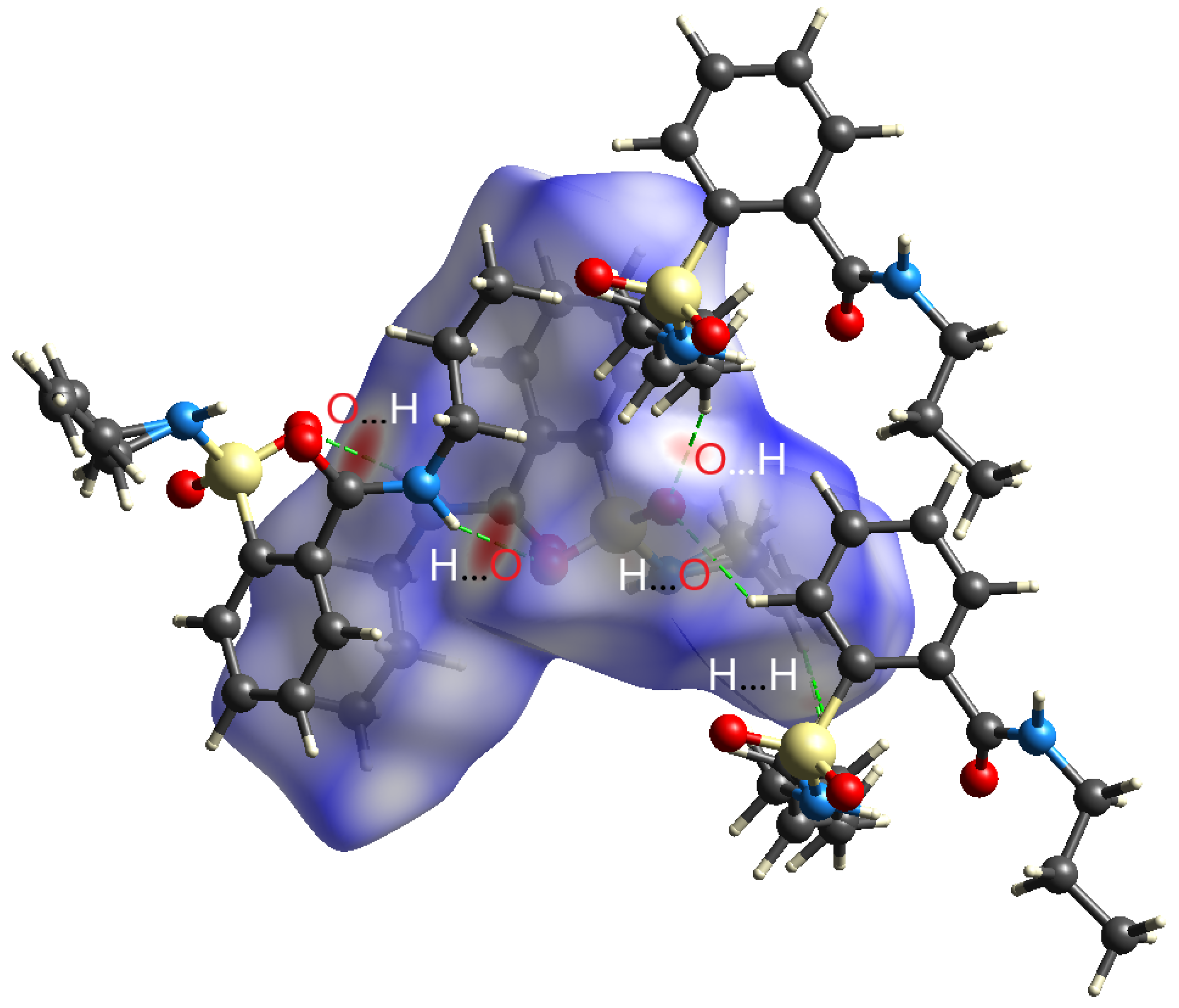
2.2. Crystal Structure Determination
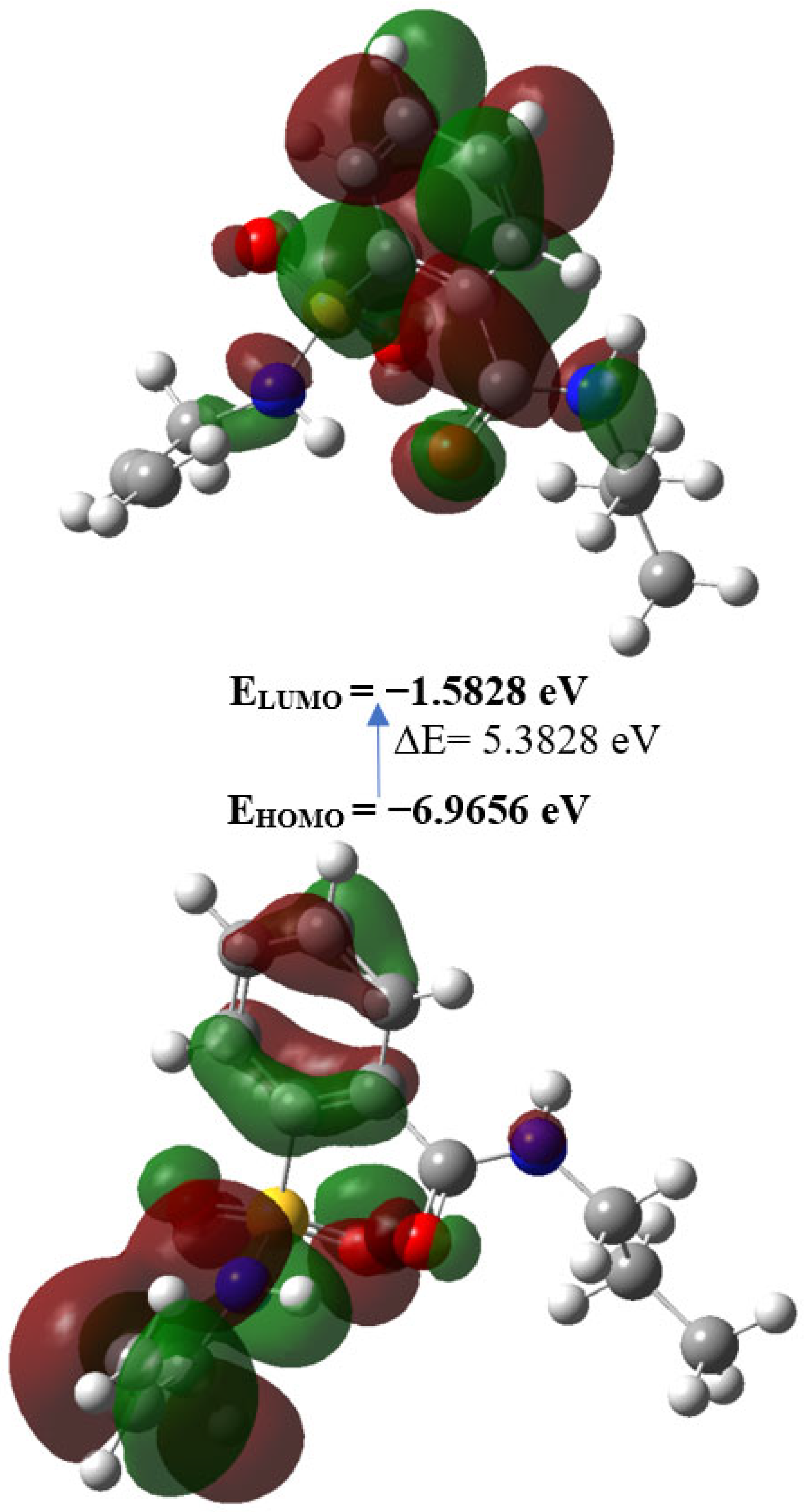
2.3. Density Functional Theory Calculations
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of Compound 4
3.3. X-ray Data of Crystal Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mustafa, M.; Winum, J.Y. The importance of sulfur-containing motifs in drug design and discovery. Expert Opin. Drug Discov. 2022, 17, 501. [Google Scholar] [CrossRef]
- Giovannuzzi, S.; D’Ambrosio, M.; Luceri, C.; Osman, S.M.; Pallecchi, M.; Bartolucci, G.; Nocentini, A.; Supuran, C.T. Aromatic sulfonamides including a sulfonic acid tail: New membrane impermeant carbonic anhydrase inhibitors for targeting selectively the cancer-associated isoforms. Int. J. Mol. Sci. 2022, 23, 461. [Google Scholar] [CrossRef]
- Ferraroni, M.; Angeli, A.; Pinteala, M.; Supuran, C.T. Sulfonamide diuretic azosemide as an efficient carbonic anhydrase inhibitor. J. Mol. Struct. 2022, 1268, 133672. [Google Scholar] [CrossRef]
- Rubab, L.; Afroz, S.; Ahmad, S.; Hussain, S.; Nawaz, I.; Irfan, A.; Batool, F.; Kotwica-Mojzych, K.; Mojzych, M. An update on synthesis of coumarin sulfonamides as enzyme inhibitors and anticancer agents. Molecules 2022, 27, 1604. [Google Scholar] [CrossRef] [PubMed]
- Ghomashi, R.; Ghomashi, S.; Aghaei, H.; Massah, A.R. Recent Advances in Biological Active Sulfonamide based Hybrid Compounds Part A: Two-Component Sulfonamide Hybrids. Curr. Med. Chem. 2023, 30, 407. [Google Scholar] [PubMed]
- Moskalik, M.Y. Sulfonamides with Heterocyclic Periphery as Antiviral Agents. Molecules 2023, 28, 51. [Google Scholar] [CrossRef]
- Deng, Z.; Sun, H.; Bheemanaboina, R.R.Y.; Luo, Y.; Zhou, C.H. Natural aloe emodin-hybridized sulfonamide aminophosphates as novel potential membrane-perturbing and DNA-intercalating agents against Enterococcus faecalis. Bioorg. Med. Chem. 2022, 64, 128695. [Google Scholar] [CrossRef]
- Portela, M.B.; Barboza, C.M.; da Silva, E.M.; de Moraes, D.C.; Simão, R.A.; de Souza, C.R.; Da Silva Cardoso, V.; Ferreria-Pereria, A.; Vermelho, A.B.; Supuran, C.T. Dentine biomodification by sulphonamides pre-treatment: Bond strength, proteolytic inhibition, and antimicrobial activity. J. Enzym. Inhib. Med. Chem. 2023, 38, 319. [Google Scholar] [CrossRef]
- Köksal, Z.; Kalin, R.; Camadan, Y.; Usanmaz, H.; Almaz, Z.; Gülçin, I.; Gokcen, T.; Ceyhan Goren, A.; Ozdemir, H. Secondary sulfonamides as effective lactoperoxidase inhibitors. Molecules 2017, 22, 793. [Google Scholar] [CrossRef] [Green Version]
- Azevedo-Barbosa, H.; Dias, D.F.; Franco, L.L.; Hawkes, J.A.; Carvalho, D.T. From antibacterial to antitumour agents: A brief review on the chemical and medicinal aspects of sulfonamides. Mini-Rev. Med. Chem. 2020, 20, 2052–2066. [Google Scholar] [CrossRef]
- Mondal, S.; Malakar, S. Synthesis of sulfonamide and their synthetic and therapeutic applications: Recent advances. Tetrahedron 2020, 76, 131662. [Google Scholar] [CrossRef]
- EL Mahmoudi, A.; Tachallait, H.; Moutaoukil, Z.; Arshad, S.; Karrouchi, K.; Benhida, R.; Bougrin, K. Ultrasound-Assisted Green Synthesis of 3, 5-Disubstituted Isoxazole Secondary Sulfonamides via One-Pot Five-Component Reaction using CaCl2/K2CO3 as Pre-Catalyst in Water. ChemistrySelect 2022, 7, e202203072. [Google Scholar] [CrossRef]
- El Mahmoudi, A.; El Masaoudi, H.; Tachallait, H.; Talha, A.; Arshad, S.; Benhida, R.; Jaber, B.; Benaissa, M.; Bougrin, K. Selective silver (I)-catalyzed four-component gram-scale synthesis of novel 1,4-disubstituted 1,2,3-triazole-sulfonamides under heterogeneous catalysis and microwave irradiation in water. Results Chem. 2022, 4, 100552. [Google Scholar] [CrossRef]
- El Mahmoudi, A.; Chkirate, K.; Tachallait, H.; Van Meervelt, L.; Bougrin, K. 2-((3-(4-Methoxyphenyl)-4,5-dihydroisoxazol-5-yl)methyl)benzo[d]isothiazol-3(2H)-one1,1-dioxide. Molbank 2022, 2022, M1488. [Google Scholar] [CrossRef]
- Bruker. APEX4, SAINT & SHELXTL; Bruker AXS LLC: Madison, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chkirate, K.; Azgaou, K.; Elmsellem, H.; El Ibrahimi, B.; Sebbar, N.K.; Anouar, E.H.; Benmessaoud, M.; El Hajjaji, S.; Essassi, E.M. Corrosion Inhibition Potential of 2-[(5-methylpyrazol-3-yl)methyl]benzimidazole against carbon steel corrosion in 1M HCl solution: Combining Experimental and Theoretical Studies. J. Mol. Liq. 2021, 321, 114750. [Google Scholar] [CrossRef]
- Bouzian, Y.; Sert, Y.; Karrouchi, K.; Van Meervelt, L.; Chkirate, K.; Mahi, L.; Hamou Ahabchane, N.; Talbaoui, A.; Essassi, E.M. Synthesis, spectroscopic characterization, DFT, molecular docking and in vitro antibacterial potential of novel quinoline derivatives. J. Mol. Struct. 2021, 1246, 131217. [Google Scholar] [CrossRef]
- Chkirate, K.; Akachar, J.; Hni, B.; Hökelek, T.; Anouar, E.H.; Talbaoui, A.; Mague, J.T.; Sebbar, N.K.; Ibrahimi, A.; Essassi, E.M. Synthesis, spectroscopic characterization, crystal structure, DFT, ESI-MS studies, molecular docking and in vitro antibacterial activity of 1,5-benzodiazepin-2-one derivatives. J. Mol. Struct. 2022, 1247, 131188. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2012. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N1—H1···O3 | 0.853 (18) | 2.108 (18) | 2.8759 (14) | 149.5 (15) |
| N2—H2A···O2 i | 0.855 (16) | 2.175 (16) | 2.9818 (13) | 157.4 (14) |
| C9—H9B···O1 ii | 0.95 | 2.55 | 3.480 (6) | 165 |
| C12—H12A···Cg1 i | 0.99 | 2.76 | 3.6902 (15) | 156 |
| X-ray | B3LYP/6–311 G(d,p) | |
|---|---|---|
| S1-N1 | 1.6238 (10) | 1.6711 |
| S1-C1 | 1.7769 (11) | 1.8111 |
| S1-O1 | 1.4327 (9) | 1.4619 |
| N1-C7 | 1.4816 (16) | 1.4674 |
| C10-O3 | 1.2353 (14) | 1.2265 |
| C10-N2 | 1.3335 (15) | 1.3557 |
| N2-C11 | 1.4614 (14) | 1.4616 |
| S1-O2 | 1.4382 (8) | 1.4651 |
| C1-S1-O1 | 107.19 (5) | 108.2414 |
| O1-S1-N1 | 107.83 (5) | 106.6917 |
| S1-N1-C7 | 116.25 (9) | 118.1819 |
| N1-C7-C8 | 108.95 (13) | 111.869 |
| C6-C1-S1 | 118.00 (9) | 116.7889 |
| C2-C1-S1 | 121.07 (8) | 121.8659 |
| C2-C10-O3 | 120.14 (10) | 121.6204 |
| C2-C10-N2 | 116.03 (10) | 114.9641 |
| C10-N2-C11 | 121.61 (10) | 121.9677 |
| N2-C11-C12 | 113.69 (10) | 113.0569 |
| C1-S1-O2 | 107.73 (5) | 106.7981 |
| O3-C10-N2 | 123.75 (10) | 123.3388 |
| Molecular Energy | Title Compound |
|---|---|
| Total energy TE (eV) | −33,735.8573 |
| EHOMO (eV) | −6.9656 |
| ELUMO (eV) | −1.5828 |
| Gap, ΔE (eV) | 5.3828 |
| Dipole moment, µ (Debye) | 4.6564 |
| Ionization potential, I (eV) | 6.9656 |
| Electron affinity, A | 1.5828 |
| Electronegativity, χ | 4.2742 |
| Hardness, η | 2.6914 |
| Electrophilicity index ω | 3.3939 |
| Softness, σ | 0.3716 |
| Fraction of electron transferred, ΔN | 0.5064 |
| Crystal Data | |
|---|---|
| Empirical formula | C13H18N2O3S |
| Formula weight | 282.35 |
| Temperature/K | 150 |
| Crystal system, space group | Monoclinic, P21/c |
| a, b, c (Å) | 8.2659 (3), 21.4034 (8), 8.3357 (3) |
| β (°) | 106.366 (1) |
| Volume (Å3) | 1414.98 (9) |
| Z | 4 |
| Radiation type | Cu Kα |
| µ (mm−1) | 2.09 |
| Crystal size (mm) | 0.23 × 0.13 × 0.13 |
| Data collection | |
| Diffractometer | Bruker D8 Venture Photon 3 CPAD |
| Absorption correction | Multi-scan SadabsBruker, (Bruker, Karlsruher, Germany) [28] |
| Tmin, Tmax | 0.72, 0.78 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 44,247, 2834, 2811 |
| Rint | 0.024 |
| (sin θ/λ)max (Å−1) | 0.625 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.029, 0.075, 1.04 |
| No. of reflections | 2834 |
| No. of parameters | 191 |
| No. of restraints | 4 |
| H atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.37, −0.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El mahmoudi, A.; Chkirate, K.; Mokhi, L.; Mague, J.T.; Bougrin, K. 2-(N-allylsulfamoyl)-N-propylbenzamide. Molbank 2023, 2023, M1678. https://doi.org/10.3390/M1678
El mahmoudi A, Chkirate K, Mokhi L, Mague JT, Bougrin K. 2-(N-allylsulfamoyl)-N-propylbenzamide. Molbank. 2023; 2023(3):M1678. https://doi.org/10.3390/M1678
Chicago/Turabian StyleEl mahmoudi, Ayoub, Karim Chkirate, Loubna Mokhi, Joel T. Mague, and Khalid Bougrin. 2023. "2-(N-allylsulfamoyl)-N-propylbenzamide" Molbank 2023, no. 3: M1678. https://doi.org/10.3390/M1678