
The Role of TRP Channels in Sepsis and Colitis

Abstract
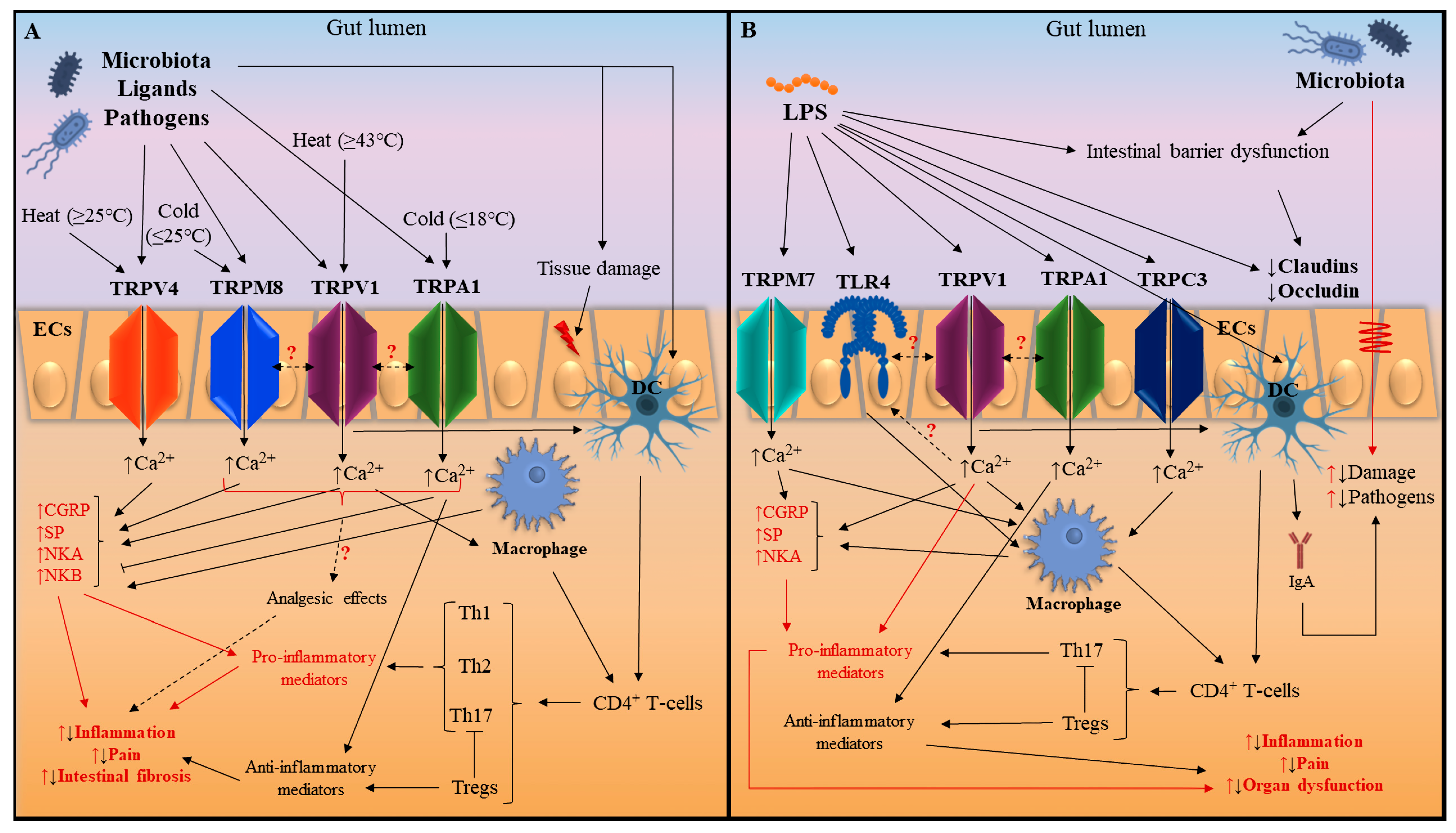
:1. Introduction
2. Modern Understanding of TRP Channels
2.1. The Role of TRP Channels in Pain
2.2. The Role of TRP Channels in Inflammation
3. Inflammatory Bowel Disease and Related Problems
3.1. TRP Channels and IBD in Humans
3.1.1. TRP Channels, Crohn’s Disease, and Ulcerative Colitis
3.1.2. TRP Channels and Intestinal Fibrosis
3.2. TRP Channels and Colitis in Animal Models
3.2.1. The TNBS-Induced Colitis Model
3.2.2. The DSS-Induced Colitis Model
3.2.3. Other Models of Colitis
4. Sepsis: A Contemporary Menace
4.1. The Gut Microenvironment in Sepsis
4.2. TRP Channels as Sensors of Bacterial Endotoxins
4.3. TRP Channels and Sepsis in Humans
4.4. TRP Channels and Sepsis in Animal Models
4.4.1. A Model of LPS-Induced Sepsis
4.4.2. Cecal Ligation and Puncture

{kind=link}
| TRP Channel | Model | Object | Confirmed and/or Expected Effects | References |
|---|---|---|---|---|
| TRPA1 | LPS-induced sepsis model | mice | Recognition of E. coli LPS independently of TLR4, possible role in the initial response to pathogens, acute neurogenic inflammation and pain | [106] |
| CLP model | mice | Protective role, regulation of the anti-inflammatory response | [128] | |
| TRPV1 | LPS-induced sepsis model | mice | LPS sensor | [112] |
| LPS-induced peritonitis | mice | Visceral pain | [114] | |
| LPS-induced peritonitis | mice | Anti-inflammatory effect on the spleen, protective effect in sepsis | [115] | |
| LPS-induced sepsis model | mice | Enhancing the antibacterial function of macrophages | [117] | |
| CLP model | mice | Protective role in bacterial infection and inflammation | [117] | |
| TRPV4 | LPS-induced sepsis model | mice | Regulation of pro-inflammatory cytokine release and LPS-stimulated macrophage phagocytosis | [118] |
| CLP model | mice | Hyperinflammatory response and mortality associated with sepsis | [127] | |
| TRPM2 | LPS-induced sepsis model | mice | Stimulation of the chemokine CXCL2 production, inflammatory and neuropathic pain | [39] |
| CLP model | mice | Regulation of pro-inflammatory cytokine production and expression of HO-1 | [129] | |
| TRPM4 | CLP model | mice | Protective effect | [131] |
| TRPM7 | LPS-induced sepsis model | mice | Regulation of TLR4 endocytosis and stimulation of inflammatory cytokine secretion associated with sepsis | [119] |
| TRPC1 | LPS-induced sepsis model | mice | Regulation of caspase-11-dependent IL-1B secretion and caspase-11-independent IL-18 secretion | [120,121] |
| TRPC3 | LPS-induced sepsis model | mice | Protective role in bacterial infections | [107] |
| TRPC4/TRPC5 functional complex | Trx+LPS model | mice | Promotion of peritoneal leukocyte accumulation and inflammatory mediator release | [122] |
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TRP | Transient receptor potential channel |
| IBD | Inflammatory bowel disease |
| CD | Crohn’s disease |
| UC | Ulcerative colitis |
| GI | Gastrointestinal tract |
| EC | Epithelial cell |
| DC | Dendritic cell |
| Th | T-helper cells |
| TLR4 | Toll-like receptor 4 |
| Tregs | Regulatory T cells |
| IgA | Immunoglobulin A |
| CGRP | Calcitonin Gene-Related Peptide |
| SP | Substance P |
| NKA | Neurokinin A |
| NKB | Neurokinin B |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| IFN | Interferon |
| NGF | Nerve growth factor |
| PAR2 | Protease-activated receptor 2 |
| APC | Antigen-presenting cell |
| TGF-β | Transforming growth factor beta |
| PDE4 | Phosphodiesterase-4 inhibitor |
| JAK | Janus Kinase Inhibitor |
| SMAD7 | Mothers against decapentaplegic homolog 7 |
| S1P | Sphingosine-1-phosphate receptor 1 |
| 4α-PDD | 4α-phorbol-12,13-didecanoate |
| siRNA | Small interfering RNA |
| TNBS | 2,4,6-Trinitrobenzene sulfonic acid |
| DRG | Dorsal root ganglion |
| NG | Nodose ganglion |
| DSS | Dextran sulfate sodium |
| WDR | Wide dynamic range |
| AITC | Allyl isothiocyanate |
| OM | Oil of mustard |
| DNBS | Dinitrobenzene sulfonic acid |
| AOM | Azoxymethane |
| SIRS | Systemic inflammatory response syndrome |
| PRRs | Pattern recognition receptors |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| NLRs | NOD-like receptors |
| RLRs | RIG-I-like receptors |
| MDSC | Myeloid-derived suppressor cell |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| AP-1 | Activating protein-1 |
| IRF-3 | Interferon regulatory factor 3 |
| CASP | Colon ascendens stent peritonitis |
| CLP | Cecal ligation and puncture |
| HO-1 | Heme oxygenase 1 |
References
- Zhang, M.; Ma, Y.; Ye, X.; Zhang, N.; Pan, L.; Wang, B. TRP (transient receptor potential) ion channel family: Structures, biological functions and therapeutic interventions for diseases. Signal Transduct. Target. Ther. 2023, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Himmel, N.J.; Cox, D.N. Transient receptor potential channels: Current perspectives on evolution, structure, function and nomenclature. Proc. R. Soc. B Biol. Sci. 2020, 287, 20201309. [Google Scholar] [CrossRef]
- Cao, E. Structural mechanisms of transient receptor potential ion channels. J. Gen. Physiol. 2020, 152, e201811998. [Google Scholar] [CrossRef] [PubMed]
- Fine, M.; Li, X.; Dang, S. Structural insights into group II TRP channels. Cell Calcium 2019, 86, 102107. [Google Scholar] [CrossRef] [PubMed]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Skerratt, S. Recent Progress in the Discovery and Development of TRPA1 Modulators. Prog. Med. Chem. 2017, 56, 81–115. [Google Scholar] [CrossRef]
- Abbas, M.A. Modulation of TRPV1 channel function by natural products in the treatment of pain. Chem. Interactions 2020, 330, 109178. [Google Scholar] [CrossRef]
- Jeong, K.-Y.; Seong, J. Neonatal capsaicin treatment in rats affects TRPV1-related noxious heat sensation and circadian body temperature rhythm. J. Neurol. Sci. 2014, 341, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.Q.; Gordon, S.E. Multimerization of Homo sapiens TRPA1 ion channel cytoplasmic domains. PLoS ONE 2019, 14, e0207835. [Google Scholar] [CrossRef]
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 Ion Channels in Inflammatory Bowel Diseases: Potential Therapeutic Targets? Pharmaceuticals 2019, 12, 48. [Google Scholar] [CrossRef]
- Vergnolle, N. TRPV4: New therapeutic target for inflammatory bowel diseases. Biochem. Pharmacol. 2014, 89, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V. Substance P and the regulation of inflammation in infections and inflammatory bowel disease. Acta Physiol. 2015, 213, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Alaimo, A.; Rubert, J. The Pivotal Role of TRP Channels in Homeostasis and Diseases throughout the Gastrointestinal Tract. Int. J. Mol. Sci. 2019, 20, 5277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Simonsen, C.; Zimova, L.; Wang, K.; Moparthi, L.; Gaudet, R.; Ekoff, M.; Nilsson, G.; Hellmich, U.A.; Vlachova, V.; et al. Cannabinoid non-cannabidiol site modulation of TRPV2 structure and function. Nat. Commun. 2022, 13, 7438. [Google Scholar] [CrossRef]
- Su, W.; Qiao, X.; Wang, W.; He, S.; Liang, K.; Hong, X. TRPV3: Structure, Diseases and Modulators. Molecules 2023, 28, 774. [Google Scholar] [CrossRef]
- Zuo, X.; Ling, J.X.; Xu, G.-Y.; Gu, J.G. Operant Behavioral Responses to Orofacial Cold Stimuli in Rats with Chronic Constrictive Trigeminal Nerve Injury: Effects of Menthol and Capsazepine. Mol. Pain 2013, 9, 28. [Google Scholar] [CrossRef]
- Tan, C.-H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nature 2016, 536, 460–463. [Google Scholar] [CrossRef]
- Jang, Y.; Cho, P.S.; Yang, Y.D.; Hwang, S.W. Nociceptive Roles of TRPM2 Ion Channel in Pathologic Pain. Mol. Neurobiol. 2018, 55, 6589–6600. [Google Scholar] [CrossRef]
- Su, S.; Yudin, Y.; Kim, N.; Tao, Y.-X.; Rohacs, T. TRPM3 Channels Play Roles in Heat Hypersensitivity and Spontaneous Pain after Nerve Injury. J. Neurosci. 2021, 41, 2457–2474. [Google Scholar] [CrossRef]
- Cortright, D.N.; Szallasi, A. TRP Channels and Pain. Curr. Pharm. Des. 2009, 15, 1736–1749. [Google Scholar] [CrossRef]
- Klimas, J.; Gorfinkel, L.; Fairbairn, N.; Amato, L.; Ahamad, K.; Nolan, S.; Simel, D.L.; Wood, E. Strategies to Identify Patient Risks of Prescription Opioid Addiction When Initiating Opioids for Pain. JAMA Netw. Open 2019, 2, e193365. [Google Scholar] [CrossRef]
- Hung, C.-Y.; Tan, C.-H. TRP Channels in Nociception and Pathological Pain. In Advances in Pain Research: Mechanisms and Modulation of Chronic Pain; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; Volume 1099, pp. 13–27. [Google Scholar] [CrossRef]
- Anderson, E.M.; Jenkins, A.C.; Caudle, R.M.; Neubert, J.K. The Effects of a Co-Application of Menthol and Capsaicin on Nociceptive Behaviors of the Rat on the Operant Orofacial Pain Assessment Device. PLoS ONE 2014, 9, e89137. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.; Chandran, P.; Hernandez, G.; Gauvin, D.M.; Mikusa, J.P.; Zhong, C.; Joshi, S.K.; Ghilardi, J.R.; Sevcik, M.A.; Fryer, R.M.; et al. Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain 2009, 142, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.; Björnsson, M.; Svensson, O.; Karlsten, R. Experiences with an adaptive design for a dose-finding study in patients with osteoarthritis. Contemp. Clin. Trials 2014, 37, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Armstrong, C. Novel therapeutic approach with PAC -14028 cream, a TRPV 1 antagonist, for patients with mild-to-moderate atopic dermatitis. Br. J. Dermatol. 2019, 180, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magieramularz, K.; Holak, T.A.; Domling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef]
- Kun, J.; Szitter, I.; Kemény, Á.; Perkecz, A.; Kereskai, L.; Pohóczky, K.; Vincze, A.; Gódi, S.; Szabó, I.; Szolcsányi, J.; et al. Upregulation of the Transient Receptor Potential Ankyrin 1 Ion Channel in the Inflamed Human and Mouse Colon and Its Protective Roles. PLoS ONE 2014, 9, e108164. [Google Scholar] [CrossRef]
- Xiao, T.; Sun, M.; Kang, J.; Zhao, C. Transient Receptor Potential Vanilloid1 (TRPV1) Channel Opens Sesame of T Cell Responses and T Cell-Mediated Inflammatory Diseases. Front. Immunol. 2022, 13, 870952. [Google Scholar] [CrossRef]
- Duo, L.; Wu, T.; Ke, Z.; Hu, L.; Wang, C.; Teng, G.; Zhang, W.; Wang, W.; Ge, Q.; Yang, Y.; et al. Gain of Function of Ion Channel TRPV1 Exacerbates Experimental Colitis by Promoting Dendritic Cell Activation. Mol. Ther. Nucleic Acids 2020, 22, 924–936. [Google Scholar] [CrossRef]
- Lapointe, T.K.; Basso, L.; Iftinca, M.C.; Flynn, R.; Chapman, K.; Dietrich, G.; Vergnolle, N.; Altier, C.; Abdullah, N.; Defaye, M.; et al. TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am. J. Physiol. Liver Physiol. 2015, 309, G87–G99. [Google Scholar] [CrossRef]
- Melnick, C.; Kaviany, M. Thermal actuation in TRPV1: Role of embedded lipids and intracellular domains. J. Theor. Biol. 2018, 444, 38–49. [Google Scholar] [CrossRef]
- Lapointe, T.K.; Altier, C. The role of TRPA1 in visceral inflammation and pain. Channels 2011, 5, 525–529. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, D.S.M.; Nassini, R.; Geppetti, P.; De Logu, F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin. Ther. Targets 2020, 24, 997–1008. [Google Scholar] [CrossRef]
- Issa, C.M.; Hambly, B.D.; Wang, Y.; Maleki, S.; Wang, W.; Fei, J.; Bao, S. TRPV2 in the Development of Experimental Colitis. Scand. J. Immunol. 2014, 80, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yamaba, R.; Inoue, K.; Utsumi, D.; Tsukahara, T.; Amagase, K.; Tominaga, M.; Kato, S. Transient receptor potential vanilloid 4 channel regulates vascular endothelial permeability during colonic inflammation in dextran sulphate sodium-induced murine colitis. Br. J. Pharmacol. 2017, 175, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.P.; Amadesi, S.; Veldhuis, N.A.; Abogadie, F.C.; Lieu, T.; Darby, W.; Liedtke, W.; Lew, M.J.; McIntyre, P.; Bunnett, N.W. Protease-activated Receptor 2 (PAR2) Protein and Transient Receptor Potential Vanilloid 4 (TRPV4) Protein Coupling Is Required for Sustained Inflammatory Signaling*. J. Biol. Chem. 2013, 288, 5790–5802. [Google Scholar] [CrossRef] [PubMed]
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 2010, 184, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 Contributes to Inflammatory and Neuropathic Pain through the Aggravation of Pronociceptive Inflammatory Responses in Mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef] [PubMed]
- Bruner, L.P.; White, A.M.; Proksell, S. Inflammatory Bowel Disease. Prim. Care Clin. Off. Pr. 2023, 50, 411–427. [Google Scholar] [CrossRef]
- Cockburn, E.; Kamal, S.; Chan, A.; Rao, V.; Liu, T.; Huang, J.Y.; Segal, J.P. Crohn’s disease: An update. Clin. Med. 2023, 23, 549–557. [Google Scholar] [CrossRef]
- Le Berre, C.; Honap, S.; Peyrin-Biroulet, L. Ulcerative colitis. Lancet 2023, 402, 571–584. [Google Scholar] [CrossRef]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Kurahara, L.-H.; Hiraishi, K. TRP channels in cardiac and intestinal fibrosis. Semin. Cell Dev. Biol. 2018, 94, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Kim, S.-H.; Kim, E.-H. The Molecular Mechanism of Transforming Growth Factor-β Signaling for Intestinal Fibrosis: A Mini-Review. Front. Pharmacol. 2019, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Di, B.; Xu, L.-L. Recent advances in the treatment of IBD: Targets, mechanisms and related therapies. Cytokine Growth Factor. Rev. 2023; 71–72, 1–12. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, F.; Peng, Y.; Yi, X.; He, Y.; Shi, Y. Research Progress of Interleukin-27 in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2023, 30, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.; Eisenstein, S. Inflammatory Bowel Disease Presentation and Diagnosis. Surg. Clin. North. Am. 2019, 99, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, M.; Jarmuż, A.; Wasilewski, A.; Sałaga, M.; Fichna, J. Role of Transient Receptor Potential Channels in Intestinal Inflammation and Visceral Pain. Inflamm. Bowel Dis. 2015, 21, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mu, J.; Zhu, M.; Mukherjee, A.; Zhang, H. Transient Receptor Potential Channels and Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Leffler, A.; Niedermirtl, F.; Babes, A.; Zimmermann, K.; Filipović, M.R.; Izydorczyk, I.; Eberhardt, M.; Kichko, T.I.; Mueller–Tribbensee, S.M.; et al. TRPA1 and Substance P Mediate Colitis in Mice. Gastroenterology 2011, 141, 1346–1358. [Google Scholar] [CrossRef]
- Du, Y.; Chen, J.; Shen, L.; Wang, B. TRP channels in inflammatory bowel disease: Potential therapeutic targets. Biochem. Pharmacol. 2022, 203, 115195. [Google Scholar] [CrossRef]
- Fischer, M.J.; Edwardson, J.M. V2A2lidating TRP channel heteromers. Temperature 2014, 1, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Bertin, S.; Aoki-Nonaka, Y.; Lee, J.; de Jong, P.R.; Kim, P.; Han, T.; Yu, T.; To, K.; Takahashi, N.; Boland, B.S.; et al. The TRPA1 ion channel is expressed in CD4+ T cells and restrains T-cell-mediated colitis through inhibition of TRPV1. Gut 2016, 66, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Becker, C.; Reeh, P.W.; Neurath, M.F. Role of sensory neurons in colitis: Increasing evidence for a neuroimmune link in the gut. Inflamm. Bowel Dis. 2011, 17, 1030–1033. [Google Scholar] [CrossRef]
- Bertin, S.; Aoki-Nonaka, Y.; de Jong, P.R.; Nohara, L.L.; Xu, H.; Stanwood, S.R.; Srikanth, S.; Lee, J.; To, K.; Abramson, L.; et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4+ T cells. Nat. Immunol. 2014, 15, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wang, Z.; Mu, J.; Zhu, M.; Zhen, Y.; Zhang, H. Upregulation of the transient receptor potential vanilloid 1 in colonic epithelium of patients with active inflammatory bowel disease. Int. J. Clin. Exp. Pathol. 2017, 10, 11335–11344. [Google Scholar] [PubMed]
- Rizopoulos, T.; Papadaki-Petrou, H.; Assimakopoulou, M. Expression Profiling of the Transient Receptor Potential Vanilloid (TRPV) Channels 1, 2, 3 and 4 in Mucosal Epithelium of Human Ulcerative Colitis. Cells 2018, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.; Jonkers, D.; Helyes, Z.; Hamer, H.; Ludidi, S.; Vanhoutvin, S.; Venema, K.; Dekker, J.; Szolcsányi, J.; et al. Alterations in mucosal neuropeptides in patients with irritable bowel syndrome and ulcerative colitis in remission: A role in pain symptom generation? Eur. J. Pain 2013, 17, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.; Yiangou, Y.; Facer, P.; Brydon, W.G.; Walters, J.R.F.; Anand, P.; Ghosh, S. Expression of the TRPV1 receptor differs in quiescent inflammatory bowel disease with or without abdominal pain. Gut 2010, 59, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.; Mokrowiecka, A.; Cygankiewicz, A.I.; Zakrzewski, P.K.; Małecka-Panas, E.; Janecka, A.; Krajewska, W.M.; Storr, M.A. Transient Receptor Potential Vanilloid 4 blockade protects against experimental colitis in mice: A new strategy for inflammatory bowel diseases treatment? Neurogastroenterol. Motil. 2012, 24, e557–e560. [Google Scholar] [CrossRef]
- D’Aldebert, E.; Cenac, N.; Rousset, P.; Martin, L.; Rolland, C.; Chapman, K.; Selves, J.; Alric, L.; Vinel, J.; Vergnolle, N. Transient Receptor Potential Vanilloid 4 Activated Inflammatory Signals by Intestinal Epithelial Cells and Colitis in Mice. Gastroenterology 2011, 140, 275–285.e3. [Google Scholar] [CrossRef]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, L.H.; Hiraishi, K.; Hu, Y.; Koga, K.; Onitsuka, M.; Doi, M.; Aoyagi, K.; Takedatsu, H.; Kojima, D.; Fujihara, Y.; et al. Activation of Myofibroblast TRPA1 by Steroids and Pirfenidone Ameliorates Fibrosis in Experimental Crohn’s Disease. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, K.; Kurahara, L.-H.; Sumiyoshi, M.; Hu, Y.-P.; Koga, K.; Onitsuka, M.; Kojima, D.; Yue, L.; Takedatsu, H.; Jian, Y.-W.; et al. Daikenchuto (Da-Jian-Zhong-Tang) ameliorates intestinal fibrosis by activating myofibroblast transient receptor potential ankyrin 1 channel. World J. Gastroenterol. 2018, 24, 4036–4053. [Google Scholar] [CrossRef] [PubMed]
- Bilsborough, J.M.; Fiorino, M.F.; Henkle, B.W. Select animal models of colitis and their value in predicting clinical efficacy of biological therapies in ulcerative colitis. Expert Opin. Drug Discov. 2020, 16, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takagi, K.; Kato, A.; Ishibashi, T.; Mori, Y.; Tashima, K.; Mitsumoto, A.; Kato, S.; Horie, S. Role of transient receptor potential melastatin 2 (TRPM2) channels in visceral nociception and hypersensitivity. Exp. Neurol. 2016, 285, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, F.; Johnson, C.; Leggit, A.; Grady, E.; Schenk, A.K.; Cevikbas, F.; Cedron, W.; Bondada, S.; Kirkwood, R.; Malone, B.; et al. Transient receptor potential ankyrin 1 mediates chronic pancreatitis pain in mice. Am. J. Physiol. Liver Physiol. 2013, 304, G1002–G1012. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, W.; De Man Joris, G.; De Schepper Heiko, U.; Bult, H.; Moreels, T.G.; Pelckmans, P.A.; De Winter Benedicte, Y. Role of TRPV1 and TRPA1 in visceral hypersensitivity to colorectal distension during experimental colitis in rats. Eur. J. Pharmacol. 2013, 698, 404–412. [Google Scholar] [CrossRef]
- Matsumoto, K.; Sugimoto, F.; Mizuno, T.; Hayashi, T.; Okamura, R.; Nishioka, T.; Yasuda, H.; Horie, S.; Kido, M.A.; Kato, S. Immunohistochemical characterization of transient receptor potential vanilloid types 2 and 1 in a trinitrobenzene sulfonic acid-induced rat colitis model with visceral hypersensitivity. Cell Tissue Res. 2022, 391, 287–303. [Google Scholar] [CrossRef]
- Yang, C.; Merlin, D. Unveiling Colitis: A Journey through the Dextran Sodium Sulfate-induced Model. Inflamm. Bowel Dis. 2024, izad312. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- King, J.W.; Bennett, A.S.W.; Wood, H.M.; Baker, C.C.; Alsaadi, H.; Topley, M.; Vanner, S.A.; Reed, D.E.; Lomax, A.E. Expression and function of transient receptor potential melastatin 3 in the spinal afferent innervation of the mouse colon. Am. J. Physiol. Liver Physiol. 2024, 326, G176–G186. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Matsumoto, K.; Tashima, K.; Nakamura, H.; Fujino, H.; Murayama, T.; Horie, S. TRPM8 has a key role in experimental colitis-induced visceral hyperalgesia in mice. Neurogastroenterol. Motil. 2014, 26, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Hyun, E.; Zhao, L.; Lapointe, T.K.; Chapman, K.; Hirota, C.L.; Ghosh, S.; McKemy, D.D.; Vergnolle, N.; Beck, P.L.; et al. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 7476–7481. [Google Scholar] [CrossRef]
- Khalil, M.; Babes, A.; Lakra, R.; Försch, S.; Reeh, P.; Wirtz, S.; Becker, C.; Neurath, M.; Engel, M. Transient receptor potential melastatin 8 ion channel in macrophages modulates colitis through a balance-shift in TNF-alpha and interleukin-10 production. Mucosal Immunol. 2016, 9, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Khalil, M.; Mueller-Tribbensee, S.M.; Becker, C.; Neuhuber, W.L.; Neurath, M.F.; Reeh, P.W. The proximodistal aggravation of colitis depends on substance P released from TRPV1-expressing sensory neurons. J. Gastroenterol. 2011, 47, 256–265. [Google Scholar] [CrossRef]
- Santoni, G.; Farfariello, V.; Liberati, S.; Morelli, M.B.; Nabissi, M.; Santoni, M.; Amantini, C. The role of transient receptor potential vanilloid type-2 ion channels in innate and adaptive immune responses. Front. Immunol. 2013, 4, 34. [Google Scholar] [CrossRef]
- Utsumi, D.; Matsumoto, K.; Tsukahara, T.; Amagase, K.; Tominaga, M.; Kato, S. Transient receptor potential vanilloid 1 and transient receptor potential ankyrin 1 contribute to the progression of colonic inflammation in dextran sulfate sodium-induced colitis in mice: Links to calcitonin gene-related peptide and substance P. J. Pharmacol. Sci. 2018, 136, 121–132. [Google Scholar] [CrossRef]
- Mitrovic, M.; Shahbazian, A.; Bock, E.; Pabst, M.A.; Holzer, P. Chemo-nociceptive signalling from the colon is enhanced by mild colitis and blocked by inhibition of transient receptor potential ankyrin 1 channels. Br. J. Pharmacol. 2010, 160, 1430–1442. [Google Scholar] [CrossRef]
- Lee, J.; Yamamoto, T.; Kuramoto, H.; Kadowaki, M. TRPV1 expressing extrinsic primary sensory neurons play a protective role in mouse oxazolone-induced colitis. Auton. Neurosci. 2012, 166, 72–76. [Google Scholar] [CrossRef]
- Kimball, E.S.; Prouty, S.P.; Pavlick, K.P.; Wallace, N.H.; Schneider, C.R.; Hornby, P.J. Stimulation of neuronal receptors, neuropeptides and cytokines during experimental oil of mustard colitis. Neurogastroenterol. Motil. 2007, 19, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Massa, F.; Sibaev, A.; Marsicano, G.; Blaudzun, H.; Storr, M.; Lutz, B. Vanilloid receptor (TRPV1)-deficient mice show increased susceptibility to dinitrobenzene sulfonic acid induced colitis. J. Mol. Med. 2005, 84, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.M.; Kaya, I.; Nazıroğlu, M. Transient receptor potential channel stimulation induced oxidative stress and apoptosis in the colon of mice with colitis-associated colon cancer: Modulator role of Sambucus ebulus L. Mol. Biol. Rep. 2022, 50, 2207–2220. [Google Scholar] [CrossRef] [PubMed]
- Srzić, I.; Adam, V.N.; Pejak, D.T. Sepsis definition: What’s new in the Treatment Guidelines. Acta Clin. Croat. 2022, 61., 67–72. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed]
- Minasyan, H. Sepsis: Mechanisms of bacterial injury to the patient. Scand. J. Trauma, Resusc. Emerg. Med. 2019, 27, 19. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-induced immunosuppression: Mechanisms, diagnosis and current treatment options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Payen, D. The gut as a hidden source of sepsis. Minerva Anestesiol. 2020, 86, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Fay, K.T.; Ford, M.L.; Coopersmith, C.M. The intestinal microenvironment in sepsis. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- Boldingh, Q.J.; de Vries, F.E.; Boermeester, M.A. Abdominal sepsis. Curr. Opin. Crit. Care 2017, 23, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yoseph, B.P.; Klingensmith, N.J.; Liang, Z.; Breed, E.R.; Burd, E.M.; Mittal, R.; Dominguez, J.A.; Petrie, B.; Ford, M.L.; Coopersmith, C.M. Mechanisms of Intestinal Barrier Dysfunction in Sepsis. Shock 2016, 46, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. New York Acad. Sci. 2012, 1258, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.J.; Yudanin, N.; Ohmura, Y.; Kubota, M.; Grinshpun, B.; Sathaliyawala, T.; Kato, T.; Lerner, H.; Shen, Y.; Farber, D.L. Spatial Map of Human T Cell Compartmentalization and Maintenance over Decades of Life. Cell 2014, 159, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Erturk-Hasdemir, D.; Ochoa-Reparaz, J.; Reinecker, H.-C.; Kasper, D.L. Plasmacytoid Dendritic Cells Mediate Anti-inflammatory Responses to a Gut Commensal Molecule via Both Innate and Adaptive Mechanisms. Cell Host Microbe 2014, 15, 413–423. [Google Scholar] [CrossRef]
- Suzuki, K. Diversified IgA-Bacteria Interaction in Gut Homeostasis. Adv. Exp. Med. Biol. 2020, 1254, 105–116. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; van der Poll, T. Immunopathophysiology of human sepsis. EBioMedicine 2022, 86, 104363. [Google Scholar] [CrossRef]
- Boonen, B.; Alpizar, Y.A.; Meseguer, V.M.; Talavera, K. TRP Channels as Sensors of Bacterial Endotoxins. Toxins 2018, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yi, F. New insights into TRP channels. Channels 2013, 8, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, V.; Alpizar, Y.A.; Luis, E.; Tajada, S.; Denlinger, B.; Fajardo, O.; Manenschijn, J.-A.; Fernández-Pena, C.; Talavera, A.; Kichko, T.; et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 2014, 5, 3125. [Google Scholar] [CrossRef]
- Casas, J.; Meana, C.; López-López, J.R.; Balsinde, J.; Balboa, M.A. Lipin-1-derived diacylglycerol activates intracellular TRPC3 which is critical for inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 8243–8260. [Google Scholar] [CrossRef]
- Murando, F.; Peloso, A.; Cobianchi, L. Experimental Abdominal Sepsis: Sticking to an Awkward but Still Useful Translational Model. Mediat. Inflamm. 2019, 2019, 8971036. [Google Scholar] [CrossRef]
- Korneev, K.V. Mouse Models of Sepsis and Septic Shock. Mol. Biol. 2019, 53, 704–717. [Google Scholar] [CrossRef]
- Dickson, K.; Lehmann, C. Inflammatory Response to Different Toxins in Experimental Sepsis Models. Int. J. Mol. Sci. 2019, 20, 4341. [Google Scholar] [CrossRef]
- Startek, J.B.; Talavera, K.; Voets, T.; Alpizar, Y.A. Differential interactions of bacterial lipopolysaccharides with lipid membranes: Implications for TRPA1-mediated chemosensation. Sci. Rep. 2018, 8, 12010. [Google Scholar] [CrossRef]
- Diogenes, A.; Ferraz, C.; Akopian, A.; Henry, M.; Hargreaves, K. LPS Sensitizes TRPV1 via Activation of TLR4 in Trigeminal Sensory Neurons. J. Dent. Res. 2011, 90, 759–764. [Google Scholar] [CrossRef]
- Devesa, I.; Planells-Cases, R.; Fernández-Ballester, G.; González-Ros, J.M.; Ferrer-Montiel, A.; Fernández-Carvajal, A. Role of the transient receptor potential vanilloid 1 in inflammation and sepsis. J. Inflamm. Res. 2011, 4, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Ishida, K.; Yoshiyama, Y.; Tanaka, S.; Kawamata, M. TRPV1 is involved in abdominal hyperalgesia in a mouse model of lipopolysaccharide-induced peritonitis and influences the immune response via peripheral noradrenergic neurons. Life Sci. 2023, 317, 121472. [Google Scholar] [CrossRef] [PubMed]
- Bodkin, J.V.; Fernandes, E.S. TRPV1 and SP: Key elements for sepsis outcome? Br. J. Pharmacol. 2013, 170, 1279–1292. [Google Scholar] [CrossRef]
- De Winter, B.Y.; Bredenoord, A.J.; Van Nassauw, L.; De Man, J.G.; De Schepper, H.U.; Timmermans, J.-P.; Pelckmans, P.A. Involvement of afferent neurons in the pathogenesis of endotoxin-induced ileus in mice: Role of CGRP and TRPV1 receptors. Eur. J. Pharmacol. 2009, 615, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.S.; Liang, L.; Smillie, S.-J.; Kaiser, F.; Purcell, R.; Rivett, D.W.; Alam, S.; Howat, S.; Collins, H.; Thompson, S.J.; et al. TRPV1 Deletion Enhances Local Inflammation and Accelerates the Onset of Systemic Inflammatory Response Syndrome. J. Immunol. 2012, 188, 5741–5751. [Google Scholar] [CrossRef] [PubMed]
- Scheraga, R.G.; Abraham, S.; Niese, K.A.; Southern, B.D.; Grove, L.M.; Hite, R.D.; McDonald, C.; Hamilton, T.A.; Olman, M.A. TRPV4 Mechanosensitive Ion Channel Regulates Lipopolysaccharide-Stimulated Macrophage Phagocytosis. J. Immunol. 2016, 196, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Schappe, M.S.; Szteyn, K.; Stremska, M.E.; Mendu, S.K.; Downs, T.K.; Seegren, P.V.; Mahoney, M.A.; Dixit, S.; Krupa, J.K.; Stipes, E.J.; et al. Chanzyme TRPM7 Mediates the Ca2+ Influx Essential for Lipopolysaccharide-Induced Toll-Like Receptor 4 Endocytosis and Macrophage Activation. Immunity 2018, 48, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Jin, M.; Desai, B.N.; Penumaka, A.; Zhu, H.; Kober, M.; Dietrich, A.; Lipinski, M.M.; Henry, T.; Clapham, D.E.; et al. Caspase-11 Controls Interleukin-1β Release through Degradation of TRPC1. Cell Rep. 2014, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Agnew, A.; Nulty, C.; Creagh, E.M. Regulation, Activation and Function of Caspase-11 during Health and Disease. Int. J. Mol. Sci. 2021, 22, 1506. [Google Scholar] [CrossRef]
- Pereira, D.M.S.; Mendes, S.J.F.; Alawi, K.; Thakore, P.; Aubdool, A.; Sousa, N.C.F.; da Silva, J.F.R.; Castro, J.A.; Pereira, I.C.P.; Silva, L.C.N.; et al. Transient Receptor Potential Canonical Channels 4 and 5 MediateEscherichia coli-Derived Thioredoxin Effects in Lipopolysaccharide-Injected Mice. Oxidative Med. Cell. Longev. 2018, 2018, 4904696. [Google Scholar] [CrossRef]
- Drechsler, S.; Osuchowski, M. Cecal Ligation and Puncture. Methods Mol. Biol. 2021, 2321, 1–8. [Google Scholar] [CrossRef]
- Herminghaus, A.; Picker, O. Colon Ascendens Stent Peritonitis (CASP). Methods Mol. Biol. 2021, 2321, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Schabbauer, G. Polymicrobial sepsis models: CLP versus CASP. Drug Discov. Today: Dis. Model. 2012, 9, e17–e21. [Google Scholar] [CrossRef]
- Wanner, S.P.; Garami, A.; Pakai, E.; Oliveira, D.L.; Gavva, N.R.; Coimbra, C.C.; Romanovsky, A.A. Aging reverses the role of the transient receptor potential vanilloid-1 channel in systemic inflammation from anti-inflammatory to proinflammatory. Cell Cycle 2012, 11, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, T.; Sonkusare, S.K.; Teuscher, C.; Poynter, M.E.; Nelson, M.T. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Sci. Rep. 2016, 6, srep33841. [Google Scholar] [CrossRef] [PubMed]
- Silverman, H.A.; Tynan, A.; Hepler, T.D.; Chang, E.H.; Gunasekaran, M.; Li, J.H.; Huerta, T.S.; Tsaava, T.; Chang, Q.; Addorisio, M.E.; et al. Transient Receptor Potential Ankyrin-1-expressing vagus nerve fibers mediate IL-1β induced hypothermia and reflex anti-inflammatory responses. Mol. Med. 2023, 29, 4. [Google Scholar] [CrossRef]
- Qian, X.; Numata, T.; Zhang, K.; Li, C.; Hou, J.; Mori, Y.; Fang, X. Transient Receptor Potential Melastatin 2 Protects Mice against Polymicrobial Sepsis by Enhancing Bacterial Clearance. Anesthesiology 2014, 121, 336–351. [Google Scholar] [CrossRef]
- Silva, R.C.M.C.; Correa, L.H.T. Heme Oxygenase 1 in Vertebrates: Friend and Foe. Cell. Biochem. Biophys. 2021, 80, 97–113. [Google Scholar] [CrossRef]
- Serafini, N.; Dahdah, A.; Barbet, G.; Demion, M.; Attout, T.; Gautier, G.; Arcos-Fajardo, M.; Souchet, H.; Jouvin, M.-H.; Vrtovsnik, F.; et al. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J. Immunol. 2012, 189, 3689–3699. [Google Scholar] [CrossRef]

| TRP Channel | Sample Source/Cell Line | Confirmed and/or Expected Effects | References |
|---|---|---|---|
| TRPA1 | Samples from patients with active and inactive forms of CD and UC | Reduced expression of several pro-inflammatory neuropeptides, cytokines, and chemokines, a decrease in inflammation | [28] |
| Colon tissue samples from patients with active forms of CD and UC | Reduced severity of chronic T-cell-mediated colitis, protective role in inflammation | [55] | |
| TRPV1 | Colonic epithelium samples from patients with active forms of CD and UC | Colitogenic responses of CD4+ T cells and intestinal inflammation | [56,57] |
| Samples from patients with UC in remission | Abdominal pain and visceral hypersensitivity | [60] | |
| TRPV4 | Colon biopsies of patients with CD and UC | Elevated intracellular Ca2+ concentrations, release of chemokines, and chronic inflammation | [61] |
| Human intestinal epithelial cell lines Caco-2 and T84 | Pro-inflammatory response, chronic inflammation | [62] | |
| Mucosal epithelial cells of patients with UC | Increased inflammation | [58] |
| TRP Channel | Model | Object | Confirmed and/or Expected Effects | References |
|---|---|---|---|---|
| TRPA1 | TNBS-induced colitis model | mice | Increased inflammation and fibrosis, acute pain | [69] |
| TNBS-induced colitis model | rat | Visceral hypersensitivity (a synergistic effect with TRPV1) | [70] | |
| DSS-induced colitis model | mice | Protective role in colitis | [28] | |
| DSS-induced colitis model | mice | Visceral hypersensitivity mediated by TRPA1 agonist AITC | [81] | |
| OM-induced colitis | mice | Increased mRNA levels of various neuropeptides and mediators associated with pain and inflammation | [83] | |
| AOM/DSS model | mice | Increased apoptotic and oxidative effects in colon cancer cells (combined effect of TRPA1, TRPM2 and TRPV1) | [85] | |
| TRPV1 | TNBS-induced colitis model | rat | Visceral hypersensitivity (a synergistic effect with TRPV2) | [71] |
| TNBS-induced colitis model | rat | Visceral hypersensitivity (a synergistic effect with TRPA1) | [70] | |
| DSS-induced colitis model | mice | Increased inflammation, increased release of CGRP and SP, visceral hypersensitivity and pain-related behavior | [30,31,78,80] | |
| Oxazolone-induced colitis | mice | Excessive neutrophil accumulation | [82] | |
| DNBS-induced colitis model | mice | Modulating of sensory pathways involved in colonic inflammation, possible protective effect | [84] | |
| AOM/DSS model | mice | Increased apoptotic and oxidative effects in colon cancer cells (combined effect of TRPA1, TRPM2 and TRPV1) | [85] | |
| TRPV2 | TNBS-induced colitis model | rat | Visceral hypersensitivity (a synergistic effect with TRPV1) | [71] |
| DSS-induced colitis model | mice | Increased inflammation | [35,79] | |
| TRPV4 | TNBS-induced colitis model | mice | Intestinal inflammation and colitis-associated pain | [61] |
| DSS-induced colitis model | mice | Pro-inflammatory effects | [36] | |
| TRPM2 | TNBS-induced colitis model | rat | Increased visceromotor reflexes caused by balloon pressure, visceral hypersensitivity | [68] |
| DSS-induced colitis model | mice | Progression of colitis through its possible implication in oxidative stress signaling | [73] | |
| AOM/DSS model | mice | Increased apoptotic and oxidative effects in colon cancer cells (combined effect of TRPA1, TRPM2 and TRPV1) | [85] | |
| TRPM3 | DSS-induced colitis model | mice | Perception of noxious stimuli in colitis, colonic hypersensitivity | [74] |
| TRPM8 | TNBS-induced colitis model | mice | Visceral hyperalgesia | [75] |
| DSS-induced colitis model | mice | Anti-inflammatory effects | [76] |
| TRP Channel | Human Cell Line | Confirmed and/or Expected Effects | References |
|---|---|---|---|
| TRPV1 | HEK293T cells | LPS sensor, impaired membrane stability, pain and inflammation associated with bacterial infection | [103] |
| TRPM2 | Human THP-1 monocytes | Regulation of LPS-induced production of pro-inflammatory cytokines IL-6, IL-8, IL-10, and TNF-α | [38] |
| TRPM3 | HEK293T cells | LPS sensor, impaired membrane stability | [103] |
| TRPM8 | HEK293T cells | LPS sensor, impaired membrane stability | [103] |
| TRPC3 | HEK293 cells (HEK-TLR4 cells) | Significant increase in the expression levels of pro-inflammatory genes PTGS2, TNFA and IL-12B, immune response to bacterial endotoxins | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvornikova, K.A.; Platonova, O.N.; Bystrova, E.Y. The Role of TRP Channels in Sepsis and Colitis. Int. J. Mol. Sci. 2024, 25, 4784. https://doi.org/10.3390/ijms25094784
Dvornikova KA, Platonova ON, Bystrova EY. The Role of TRP Channels in Sepsis and Colitis. International Journal of Molecular Sciences. 2024; 25(9):4784. https://doi.org/10.3390/ijms25094784
Chicago/Turabian StyleDvornikova, Kristina A., Olga N. Platonova, and Elena Y. Bystrova. 2024. "The Role of TRP Channels in Sepsis and Colitis" International Journal of Molecular Sciences 25, no. 9: 4784. https://doi.org/10.3390/ijms25094784