
Deciphering the Impact of Defecation Frequency on Gut Microbiome Composition and Diversity

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. General Characteristics of the Subjects
2.2. Identification of Enterotypes among All Subjects
2.3. Microbial Diversity and Structure Differences According to Defecation Frequency
2.4. Microbial Composition Difference and Relationship with Defecation Frequency
2.5. Predictive KEGG Functional Profiling
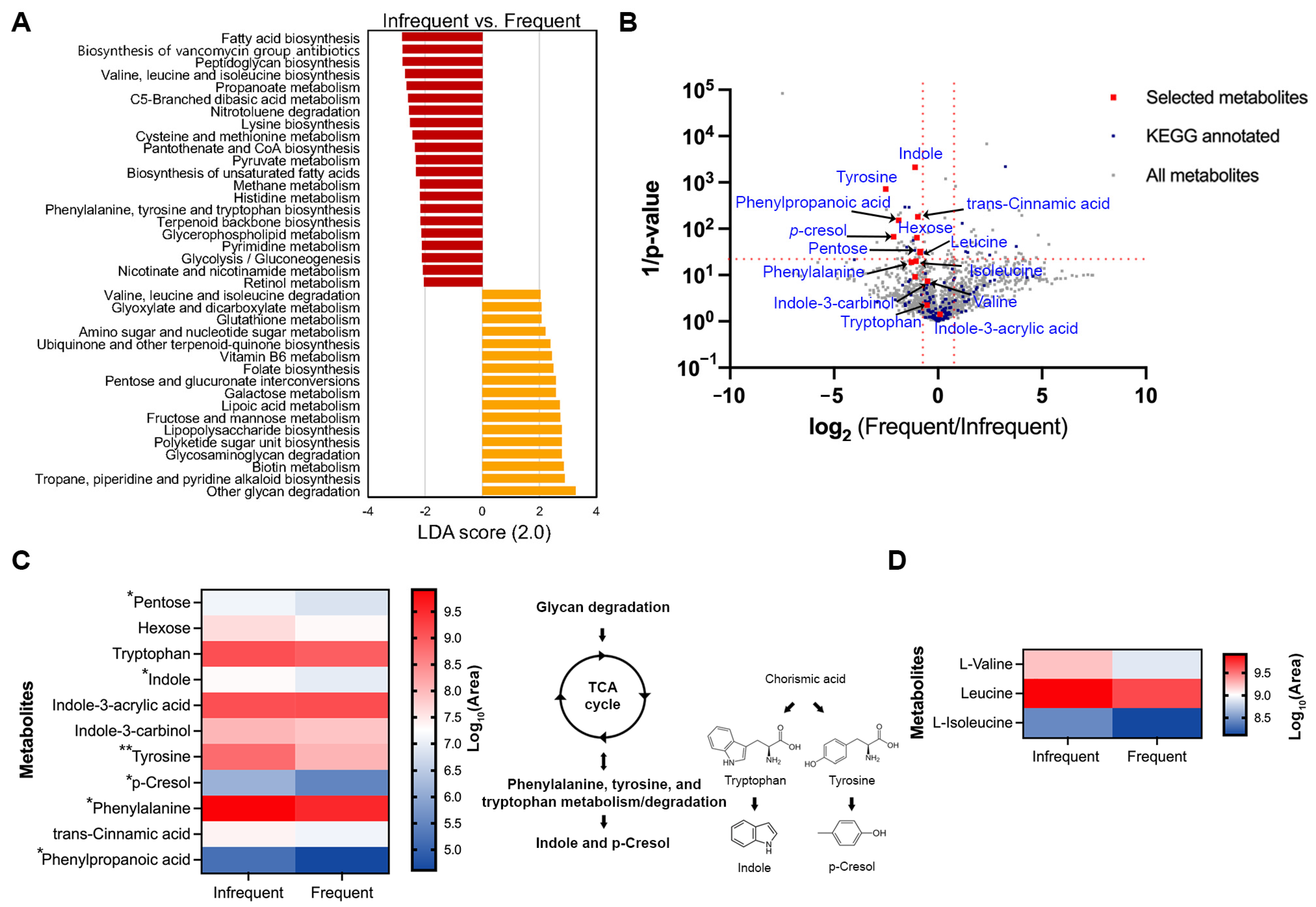
2.6. Differences in the Metabolites Related to Defecation Frequency
2.7. Random Forest Prediction
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. The 16S rRNA Amplicon Sequencing
4.3. Data Analysis
4.4. Enterotype Analysis
4.5. Predictive KEGG Function Profiling
4.6. Sample Preparation for Global Metabolomics
4.7. U-HPLC-MS/MS Conditions
4.8. Global Metabolomic Analysis
4.9. Random Forest
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 2013, 9, e1002863. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Christensen, L.; Roager, H.M.; Astrup, A.; Hjorth, M.F. Microbial enterotypes in personalized nutrition and obesity management. Am. J. Clin. Nutr. 2018, 108, 645–651. [Google Scholar] [CrossRef]
- Song, E.J.; Han, K.; Lim, T.J.; Lim, S.; Chung, M.J.; Nam, M.H.; Kim, H.; Nam, Y.D. Effect of probiotics on obesity-related markers per enterotype: A double-blind, placebo-controlled, randomized clinical trial. EPMA J. 2020, 11, 31–51. [Google Scholar] [CrossRef]
- Zou, H.; Wang, D.; Ren, H.; Cai, K.; Chen, P.; Fang, C.; Shi, Z.; Zhang, P.; Wang, J.; Yang, H.; et al. Effect of Caloric Restriction on BMI, Gut Microbiota, and Blood Amino Acid Levels in Non-Obese Adults. Nutrients 2020, 12, 631. [Google Scholar] [CrossRef]
- Lim, M.Y.; Rho, M.; Song, Y.M.; Lee, K.; Sung, J.; Ko, G. Stability of gut enterotypes in Korean monozygotic twins and their association with biomarkers and diet. Sci. Rep. 2014, 4, 7348. [Google Scholar] [CrossRef]
- Lee, S.H.; Yoon, S.H.; Jung, Y.; Kim, N.; Min, U.; Chun, J.; Choi, I. Emotional well-being and gut microbiome profiles by enterotype. Sci. Rep. 2020, 10, 20736. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Hansen, L.B.; Bahl, M.I.; Frandsen, H.L.; Carvalho, V.; Gøbel, R.J.; Dalgaard, M.D.; Plichta, D.R.; Sparholt, M.H.; Vestergaard, H. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 2016, 1, 16093. [Google Scholar] [CrossRef]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Lim, J.H.; Kang, D.; Lim, S.; Park, S.J.; Kim, J.H. Is stool frequency associated with the richness and community composition of gut microbiota? Intest. Res. 2019, 17, 419–426. [Google Scholar] [CrossRef]
- Hadizadeh, F.; Walter, S.; Belheouane, M.; Bonfiglio, F.; Heinsen, F.A.; Andreasson, A.; Agreus, L.; Engstrand, L.; Baines, J.F.; Rafter, J.; et al. Stool frequency is associated with gut microbiota composition. Gut 2017, 66, 559–560. [Google Scholar] [CrossRef]
- Asnicar, F.; Leeming, E.R.; Dimidi, E.; Mazidi, M.; Franks, P.W.; Al Khatib, H.; Valdes, A.M.; Davies, R.; Bakker, E.; Francis, L.; et al. Blue poo: Impact of gut transit time on the gut microbiome using a novel marker. Gut 2021, 70, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Dimidi, E.; Mark Scott, S.; Whelan, K. Probiotics and constipation: Mechanisms of action, evidence for effectiveness and utilisation by patients and healthcare professionals. Proc. Nutr. Soc. 2020, 79, 147–157. [Google Scholar] [CrossRef]
- Macfarlane, S.; Quigley, M.E.; Hopkins, M.J.; Newton, D.F.; Macfarlane, G.T. Polysaccharide degradation by human intestinal bacteria during growth under multi-substrate limiting conditions in a three-stage continuous culture system. Fems Microbiol. Ecol. 1998, 26, 231–243. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Cummings, J.H.; Macfarlane, S.; Gibson, G.R. Influence of retention time on degradation of pancreatic enzymes by human colonic bacteria grown in a 3-stage continuous culture system. J. Appl. Bacteriol. 1989, 67, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.H.; Sanz, Y.; Tome, D. Intestinal luminal nitrogen metabolism: Role of the gut microbiota and consequences for the host. Pharmacol. Res. 2013, 68, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Candeliere, F.; Simone, M.; Leonardi; Rossi, M.; Amaretti, A.; Raimondi, S. Indole and p-cresol in feces of healthy subjects: Concentration, kinetics, and correlation with microbiome. Front. Mol. Med. 2022, 2, 959189. [Google Scholar] [CrossRef]
- Amaretti, A.; Gozzoli, C.; Simone, M.; Raimondi, S.; Righini, L.; Perez-Brocal, V.; Garcia-Lopez, R.; Moya, A.; Rossi, M. Profiling of Protein Degraders in Cultures of Human Gut Microbiota. Front. Microbiol. 2019, 10, 2614. [Google Scholar] [CrossRef] [PubMed]
- Falconi, C.A.; Junho, C.; Fogaca-Ruiz, F.; Vernier, I.C.S.; da Cunha, R.S.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front. Physiol. 2021, 12, 686249. [Google Scholar] [CrossRef]
- Harlacher, E.; Wollenhaupt, J.; Baaten, C.; Noels, H. Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 531. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Yoshida, N.; Yamashita, T.; Osone, T.; Hosooka, T.; Shinohara, M.; Kitahama, S.; Sasaki, K.; Sasaki, D.; Yoneshiro, T.; Suzuki, T.; et al. Bacteroides spp. promotes branched-chain amino acid catabolism in brown fat and inhibits obesity. iScience 2021, 24, 103342. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat. Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Dillon, M.R.; Bolyen, E.; Kaehler, B.D.; Huttley, G.A.; Caporaso, J.G. q2-sample-classifier: Machine-learning tools for microbiome classification and regression. J. Open Res. Softw. 2018, 3, 934. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1–3 Times a Week | 4–6 Times a Week | Everyday | p Value | ||
|---|---|---|---|---|---|
| Total no. of subjects | n = 4 | n = 14 | n = 14 | - | |
| No. of enrolled subjects | n = 4 | n = 7 | n = 9 | - | |
| Sex | Male, n (%) | 2 (50.0) | 1 (14.3) | 5 (55.6) | - |
| Female, n (%) | 2 (50.0) | 6 (85.7) | 4 (44.4) | - | |
| Age, average ± SD | 24.0 ± 1.2 | 24.7 ± 1.7 | 23.9 ± 1.2 | 0.540 | |
| BMI, average ± SD | 22.1 ± 1.5 | 21.3 ± 3.0 | 21.4 ± 1.1 | 0.836 | |
| Defecation Frequency | 1–3 Times | 4–6 Times | Everyday | Total |
|---|---|---|---|---|
| No. of samples | 24 | 38 | 54 | 116 |
| No. of sequences | 507,975 | 865,445 | 1,223,190 | 2,596,610 |
| Average sequence ± SD | 21,166 ± 4922 | 22,775 ± 5060 | 22,652 ± 3901 | 22,385 ± 4569 |
| No. of features (ASVs) | 459 | 486 | 532 | 870 |
| Average features (ASVs) ± SD | 124 ± 23 | 102 ± 17 | 94 ± 21 | 103 ± 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.; Kim, S.; Lee, W.; Kim, G.; Shin, H. Deciphering the Impact of Defecation Frequency on Gut Microbiome Composition and Diversity. Int. J. Mol. Sci. 2024, 25, 4657. https://doi.org/10.3390/ijms25094657
Park G, Kim S, Lee W, Kim G, Shin H. Deciphering the Impact of Defecation Frequency on Gut Microbiome Composition and Diversity. International Journal of Molecular Sciences. 2024; 25(9):4657. https://doi.org/10.3390/ijms25094657
Chicago/Turabian StylePark, Gwoncheol, Seongok Kim, WonJune Lee, Gyungcheon Kim, and Hakdong Shin. 2024. "Deciphering the Impact of Defecation Frequency on Gut Microbiome Composition and Diversity" International Journal of Molecular Sciences 25, no. 9: 4657. https://doi.org/10.3390/ijms25094657