
Mlp4green: A Binary Classification Approach Specifically for Green Odor


Abstract
:1. Introduction
2. Results
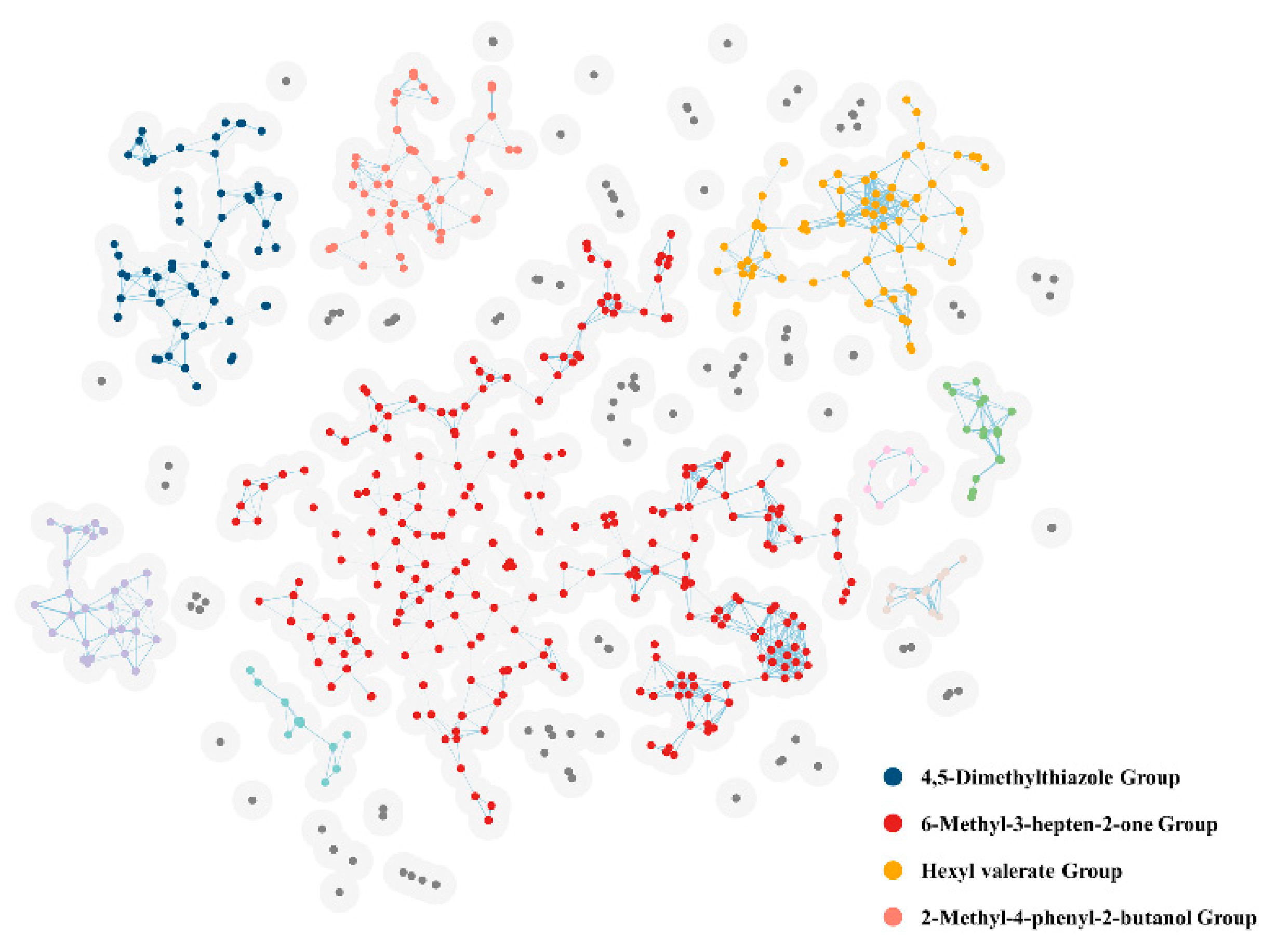
2.1. Cluster Analysis
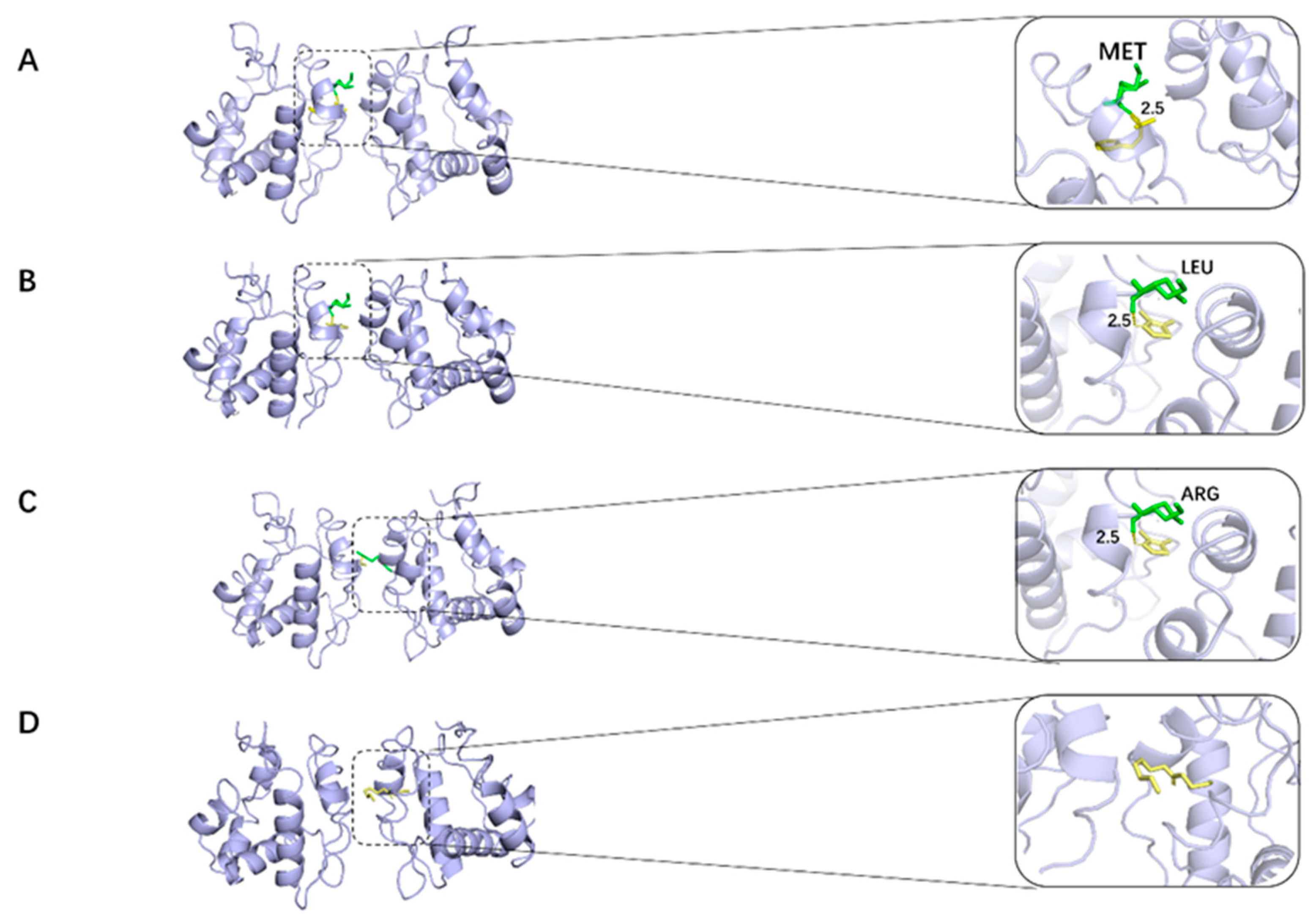
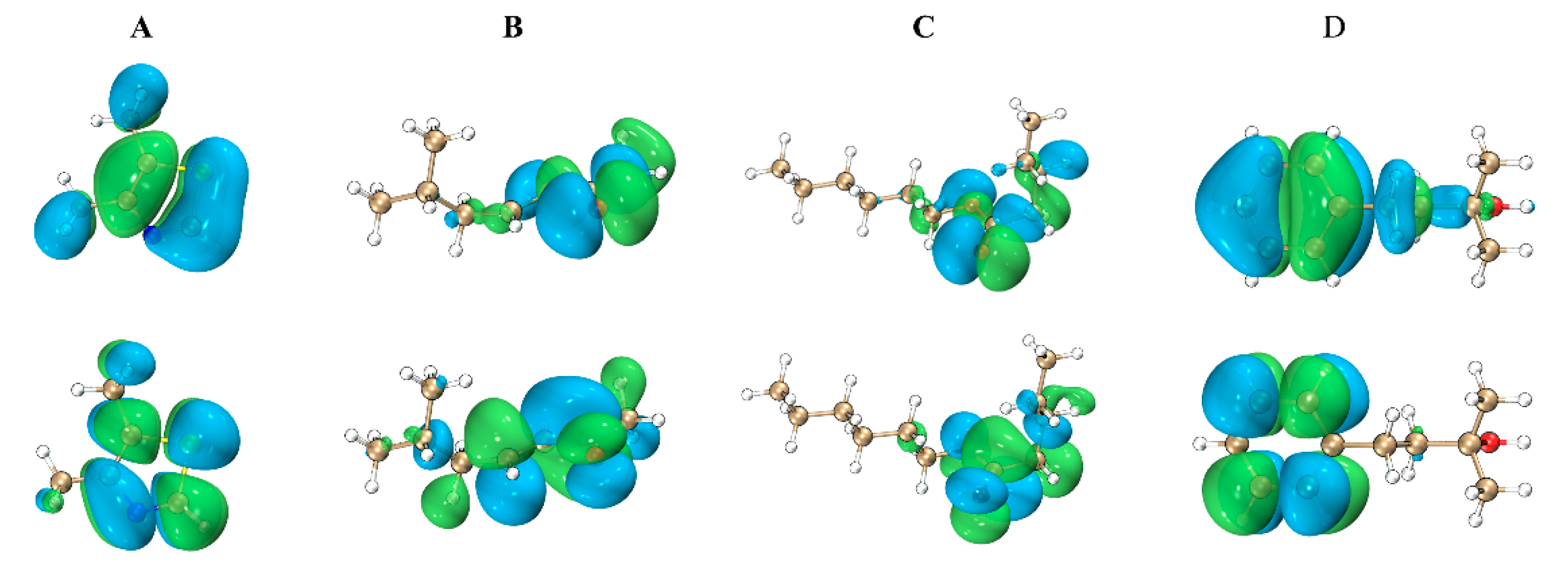
2.2. Docking Study
2.2.1. 2_Methyl_4_phenyl_2_butanol Group
2.2.2. 4,5-Dimethylthiazolel Group
2.2.3. 6-Methyl-3-hepten-2-one Group
2.2.4. Hexyl Valerate Group
2.2.5. Quantum Chemical Calculations
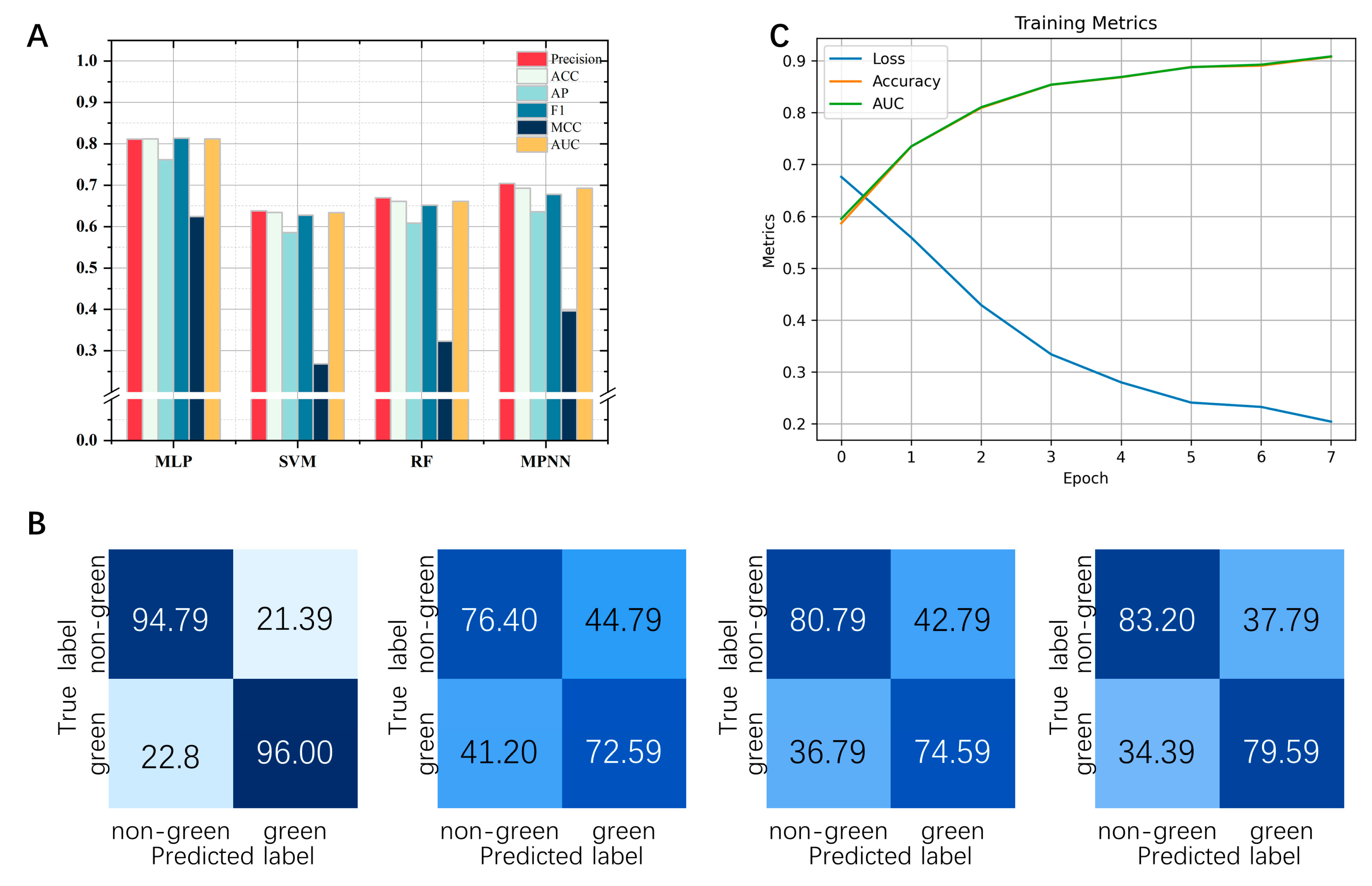
2.3. Evaluate Different Machine Learning Algorithms to Predict Green Odor
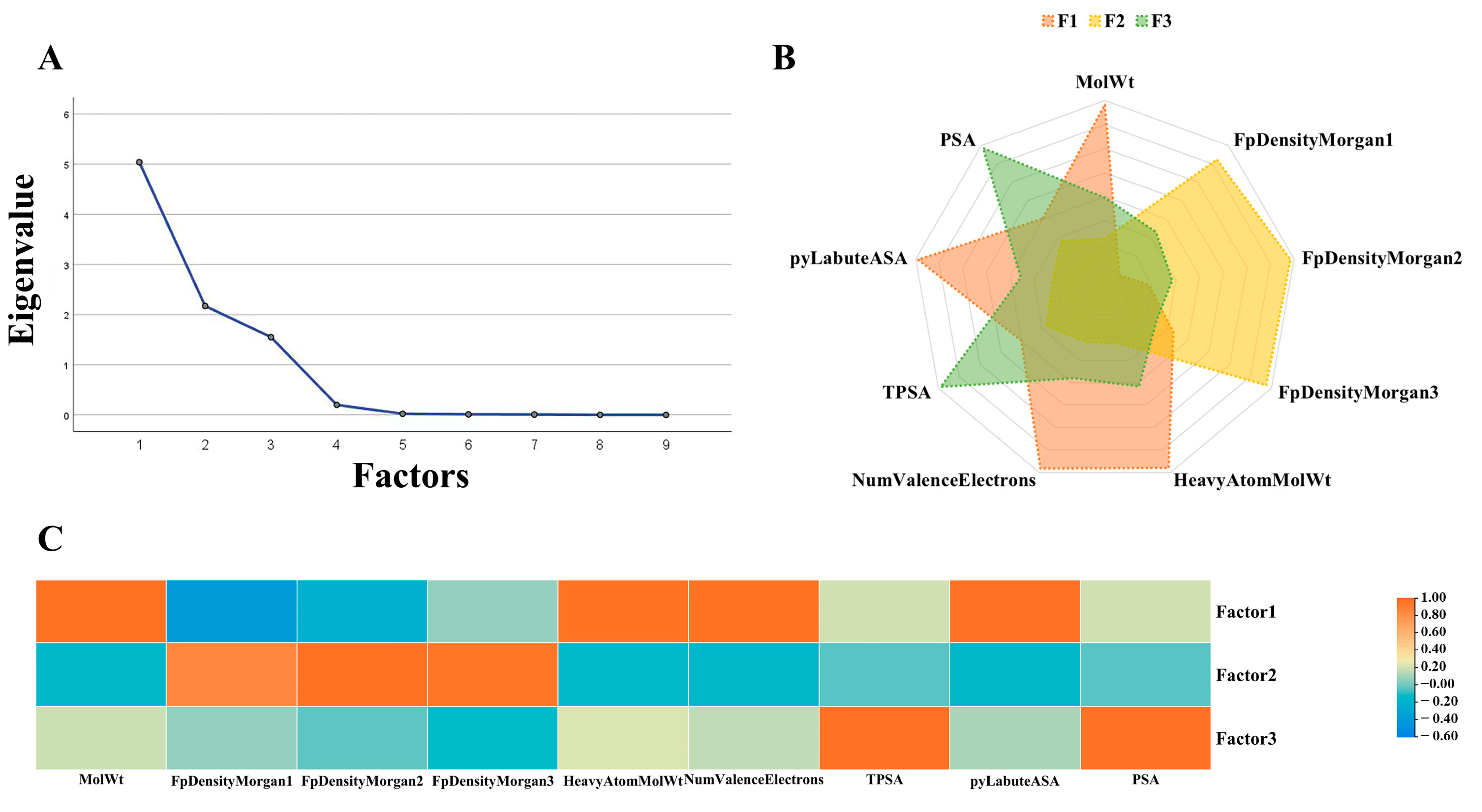
2.4. Factor Analysis
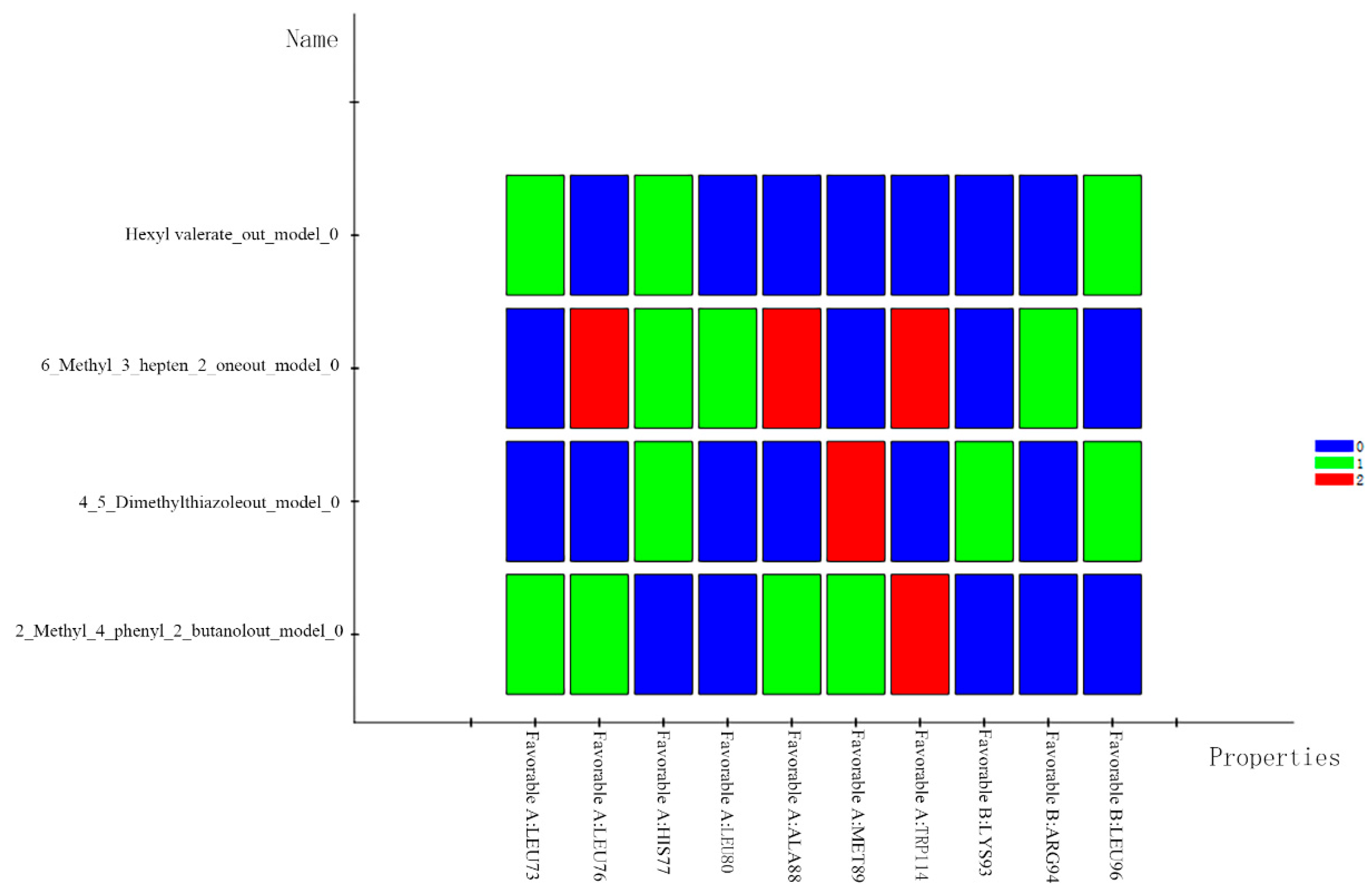
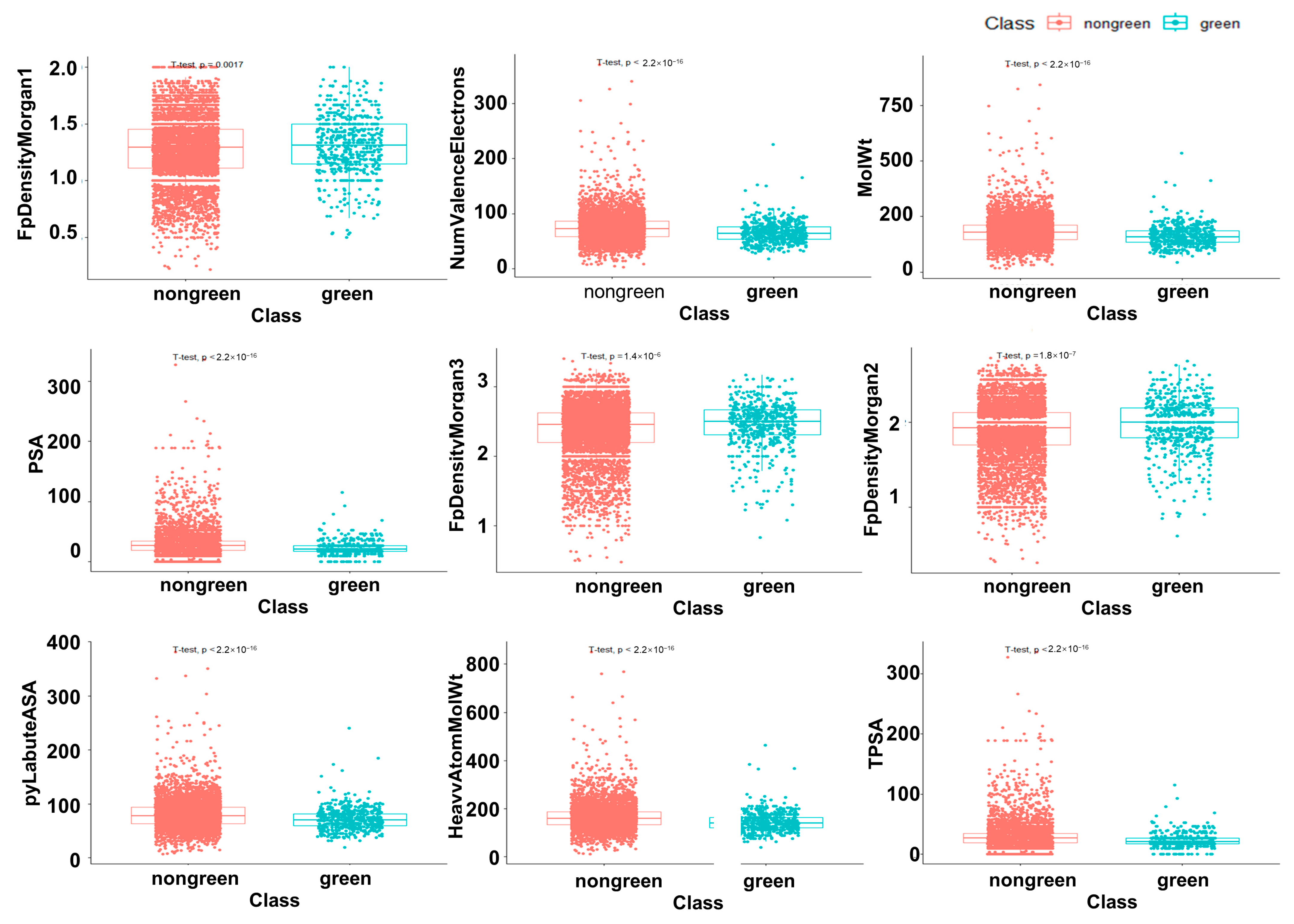
2.5. The Difference Analysis between Green and Non-Green
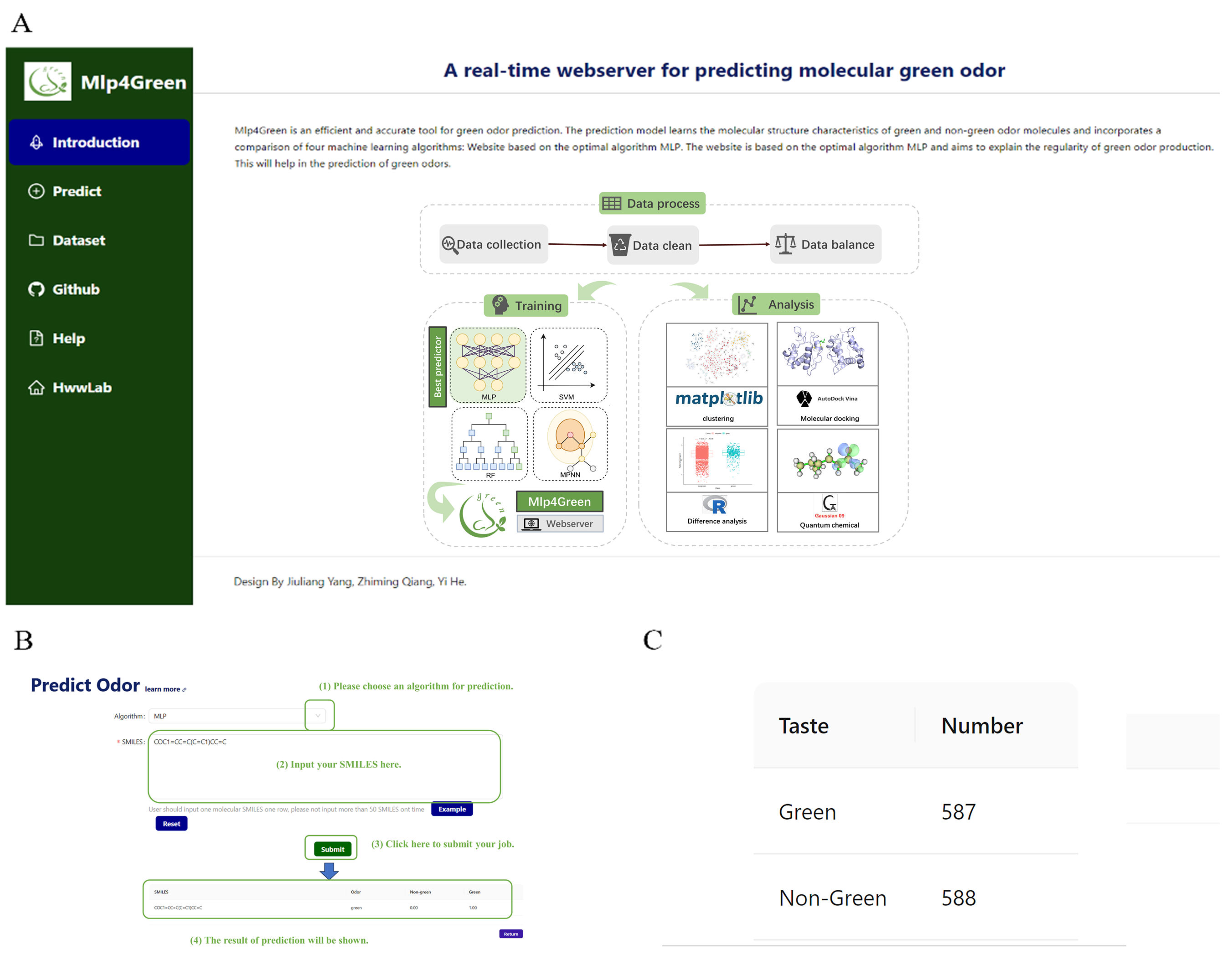
2.6. Webserver
3. Discussion
3.1. Analysis of the Chemical Properties of Four Representative Groups of Green Odor Molecules
3.2. Analysis of the Results of the Docking Study
3.3. Analysis of the Results of the Prediction Algorithms
4. Materials and Methods
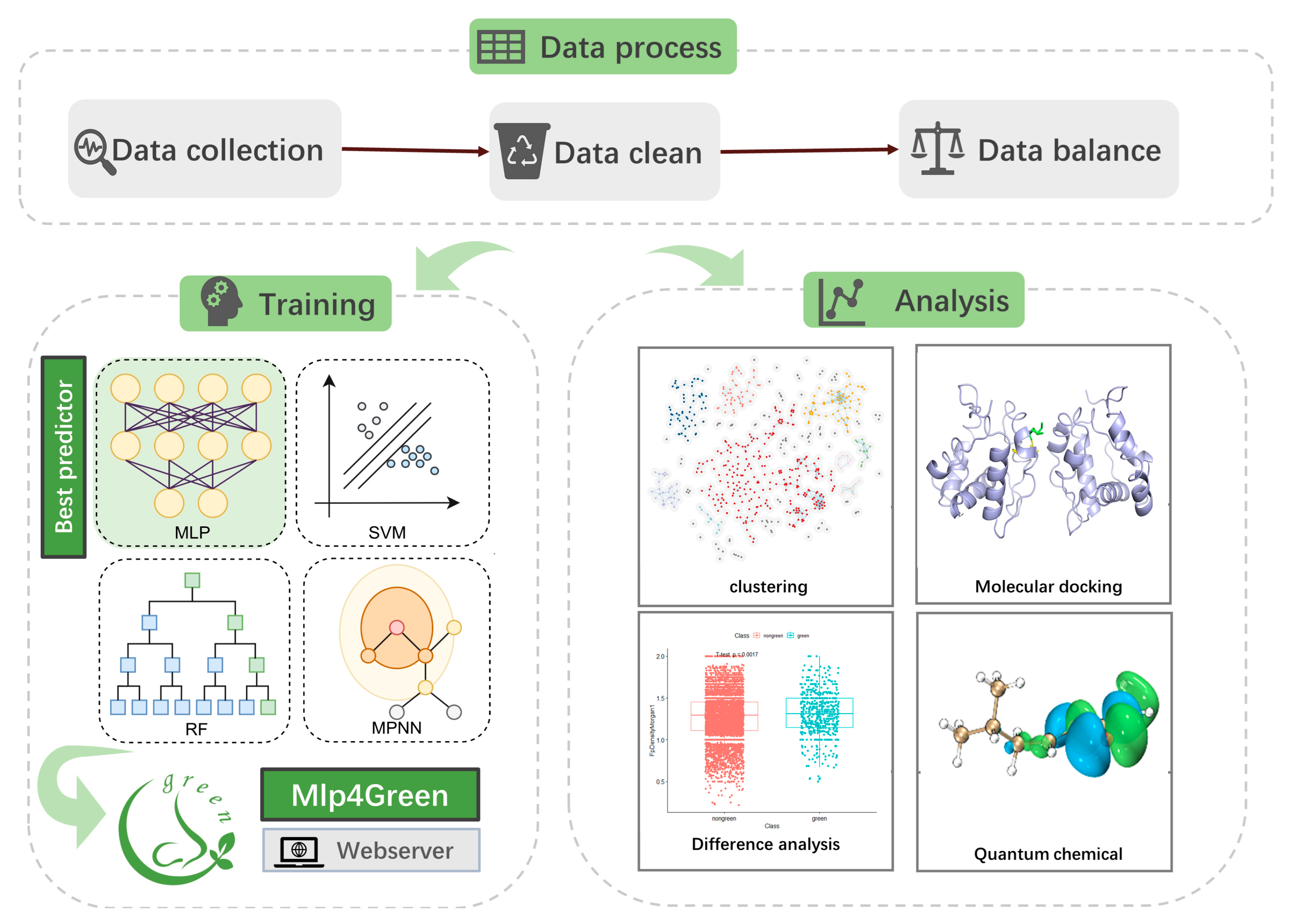
4.1. The Workflow of the Study
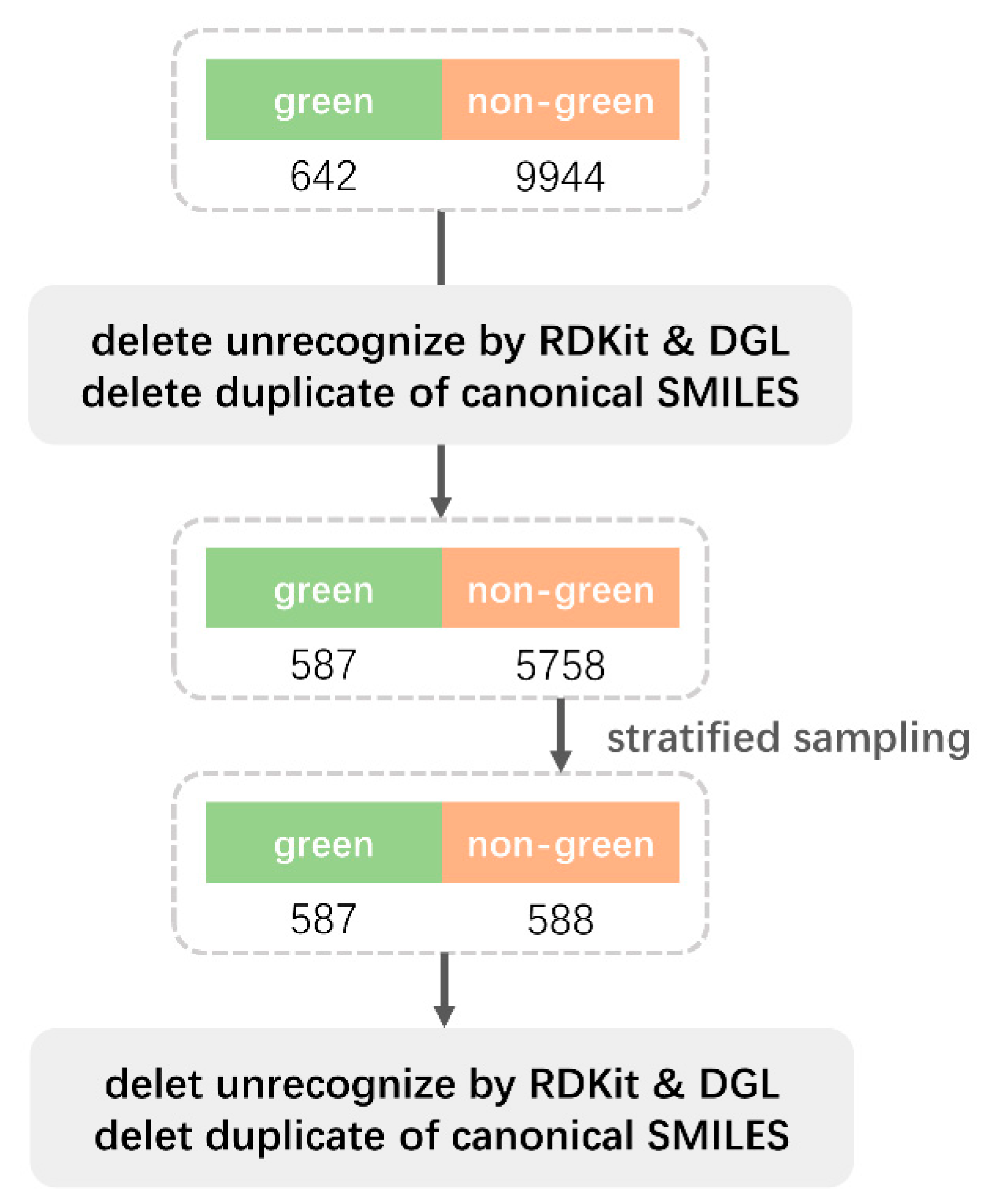
4.2. The Dataset Processing
4.3. The Models for Green Odor Prediction
- (1)
- We built MLP [22] using PyTorch 2.2.1 (https://pytorch.org/, accessed on 5 December 2023). First, using RDKit 2023.9.5 (https://www.rdkit.org/, accessed on 3 December 2023), the molecules in the dataset were transformed into morgan fingerprints with a radius of 2 and a length of 2048 bits. The batch size was set to 1/16 of the total number of molecules. Subsequently, the fingerprint feature was fed into an MLP, which had two neurons in the output layer, 256 neurons in the hidden layer, and 256 neurons in the input layer. A 0.1 dropout and a ReLU activation function were present between each layer. When the number of times the loss no longer accumulated to seven, the early stop method was utilized to end the training. The trainer employed the cross-entropy loss function, the Adam optimizer, and a learning rate of 0.001.
- (2)
- SVM [23] was built using scikit-learn 1.4.1 (https://scikit-learn.org/, accessed on 1 December 2023). The SVM uses the same input as the MLP mentioned earlier. A 5-fold cross-validated grid search technique was used to identify the SVM’s ideal parameters. The optimal parameters were “C”: 1, “gamma”: 0.1, “kernel”: “rbf”, and “probability”: True.
- (3)
- The RF model [24] uses the same input as the SVM mentioned previously. A 5-fold cross-validated grid search technique was used to identify the RF’s ideal parameters. The optimal parameters were “max_depth”: 6, “max_features”: “log2”, “min_samples_leaf”: 50, “min_samples_split”: 2, “n_estimators”: 100, and “probability”: True.
- (4)
- Using DGL-LifeSci 0.3.1 (https://lifesci.dgl.ai/, accessed on 6 December 2023), we constructed the MPNN [25]. The DGL graphs of the molecules were the MPNN’s input, and a batch size of 1/16 of the total number of molecules was selected. Using canonical atom and bond featurization, node feature and edge feature embedding produced 74 one-hot coding features for atoms and 12 one-hot coding features for bonds. The MPNN’s node output dimension and edge output dimension were both set to 64 and 128 correspondingly, with the remaining parameters remaining at their factory settings. The training parameters, similar to MLP, include early stopping, learning rate, loss function, and optimizer.
4.4. Performance Evaluation
4.5. Clustering of the Green Odor Molecules
4.6. Chemical Space Network Mapping
4.7. Factor Analysis and Difference Analysis
4.8. Dice Coefficients to Compute Morgan Fingerprint Similarity
4.9. Molecular Docking and Quantum Chemistry Calculations
4.10. Webserver
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salas, J.J.; Sánchez, C.; García-González, D.L.; Aparicio, R. Impact of the suppression of lipoxygenase and hydroperoxide lyase on the quality of the green odor in green leaves. J. Agric. Food Chem. 2005, 53, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, A. The fresh green odor emitted by plants. Food Rev. Int. 1996, 12, 303–350. [Google Scholar] [CrossRef]
- Yamate, Y.; Orita, K.; Kobayashi, H.; Hiramoto, K. Ameliorative effect of green odor against UVB-induced immunosuppression of contact hypersensitivity. Dermatol. Ther. 2020, 33, e13848. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hayashida, S.; Kaneda, Y.; Takenaga, M.; Tamagawa, Y.; Tsuji, S.; Hatanaka, A. Green odor attenuates a cold pressor test-induced cardiovascular response in healthy adults. Biopsychosoc. Med. 2008, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Fujihara, M.; Murakami, E.; Miyoshi, M.; Tanaka, Y.; Koba, S.; Tachibana, H. Green odor and depressive-like state in rats: Toward an evidence-based alternative medicine? Behav. Brain Res. 2011, 224, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Ueki, S.; Miyoshi, M.; Watanabe, T. “Green odor” inhalation by stressed rat dams reduces behavioral and neuroendocrine signs of prenatal stress in the offspring. Horm. Behav. 2010, 58, 264–272. [Google Scholar] [CrossRef]
- Sano, K.; Tsuda, Y.; Sugano, H.; Aou, S.; Hatanaka, A. Concentration effects of green odor on event-related potential (P300) and pleasantness. Chem. Senses 2002, 27, 225–230. [Google Scholar] [CrossRef]
- Watanabe, Y.; Sasabe, T.; Yamaguti, K.; Kobayashi, M.; Yamamoto, S.; Kuratsune, H.; Sano, K.; Hatanaka, A.; Tsukada, H.; Onoe, H. Prevention and/or recovery effects by green odor(s) on fatigue and green-odor-responsible brain regions as revealed by PET. Chem. Senses 2005, 30 (Suppl. S1), i268–i269. [Google Scholar] [CrossRef]
- Nakashima, T.; Akamatsu, M.; Hatanaka, A.; Kiyohara, T. Attenuation of stress-induced elevations in plasma ACTH level and body temperature in rats by green odor. Physiol. Behav. 2004, 80, 481–488. [Google Scholar] [CrossRef]
- Nikaido, Y.; Yamada, J.; Migita, K.; Shiba, Y.; Furukawa, T.; Nakashima, T.; Ueno, S. cis-3-Hexenol and trans-2-hexenal mixture prevents development of PTSD-like phenotype in rats. Behav. Brain Res. 2016, 297, 251–258. [Google Scholar] [CrossRef]
- Nakamura, S.; Hatanaka, A. Green-leaf-derived C6-aroma compounds with potent antibacterial action that act on both Gram-negative and Gram-positive bacteria. J. Agric. Food Chem. 2002, 50, 7639–7644. [Google Scholar] [CrossRef] [PubMed]
- Poivet, E.; Tahirova, N.; Peterlin, Z.; Xu, L.; Zou, D.J.; Acree, T.; Firestein, S. Functional odor classification through a medicinal chemistry approach. Sci. Adv. 2018, 4, eaao6086. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.; Ramanathan, V. Predicting odor from molecular structure: A multi-label classification approach. Sci. Rep. 2022, 12, 13863. [Google Scholar] [CrossRef]
- Zheng, X.; Tomiura, Y.; Hayashi, K. Investigation of the structure-odor relationship using a Transformer model. J. Cheminform. 2022, 14, 88. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, R.; Ranjta, S.; Varadwaj, P.K. SMILES to Smell: Decoding the Structure-Odor Relationship of Chemical Compounds Using the Deep Neural Network Approach. J. Chem. Inf. Model. 2021, 61, 676–688. [Google Scholar] [CrossRef]
- Lötsch, J.; Kringel, D.; Hummel, T. Machine Learning in Human Olfactory Research. Chem. Senses 2019, 44, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bo, W.; Yu, Y.; He, R.; Qin, D.; Zheng, X.; Wang, Y.; Ding, B.; Liang, G. Insight into the Structure-Odor Relationship of Molecules: A Computational Study Based on Deep Learning. Foods 2022, 11, 2033. [Google Scholar] [CrossRef]
- Tsitsanou, K.E.; Thireou, T.; Drakou, C.E.; Koussis, K.; Keramioti, M.V.; Leonidas, D.D.; Eliopoulos, E.; Iatrou, K.; Zographos, S.E. Anopheles gambiae odorant binding protein crystal complex with the synthetic repellent DEET: Implications for structure-based design of novel mosquito repellents. Cell Mol. Life Sci. 2012, 69, 283–297. [Google Scholar] [CrossRef]
- Pelosi, P.; Iovinella, I.; Zhu, J.; Wang, G.; Dani, F.R. Beyond chemoreception: Diverse tasks of soluble olfactory proteins in insects. Biol. Rev. Camb. Philos. Soc. 2018, 93, 184–200. [Google Scholar] [CrossRef]
- Jia, C.; Mohamed, A.; Cattaneo, A.M.; Huang, X.; Keyhani, N.O.; Gu, M.; Zang, L.; Zhang, W. Odorant-Binding Proteins and Chemosensory Proteins in Spodoptera frugiperda: From Genome-Wide Identification and Developmental Stage-Related Expression Analysis to the Perception of Host Plant Odors, Sex Pheromones, and Insecticides. Int. J. Mol. Sci. 2023, 24, 5595. [Google Scholar] [CrossRef]
- Kou, X.; Shi, P.; Gao, C.; Ma, P.; Xing, H.; Ke, Q.; Zhang, D. Data-Driven Elucidation of Flavor Chemistry. J. Agric. Food Chem. 2023, 71, 6789–6802. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.K.; Sivakumar, S.; Wang, X.; Ulissi, Z.W. Cluster-MLP: An Active Learning Genetic Algorithm Framework for Accelerated Discovery of Global Minimum Configurations of Pure and Alloyed Nanoclusters. J. Chem. Inf. Model. 2023, 63, 6192–6197. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fang, Z.; Lu, S.; Gao, C. Efficacy of Regularized Multitask Learning Based on SVM Models. IEEE Trans. Cybern. 2022, 54, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, M.A.; Tanveer, M.; Suganthan, P.N.; Snasel, V. Oblique and rotation double random forest. Neural Netw. 2022, 153, 496–517. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Mayhew, E.J.; Sanchez-Lengeling, B.; Wei, J.N.; Qian, W.W.; Little, K.A.; Andres, M.; Nguyen, B.B.; Moloy, T.; Yasonik, J.; et al. A principal odor map unifies diverse tasks in olfactory perception. Science 2023, 381, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ranson, J.M.; Everson, R.; Llewellyn, D.J. Performance of Machine Learning Algorithms for Predicting Progression to Dementia in Memory Clinic Patients. JAMA Netw. Open 2021, 4, e2136553. [Google Scholar] [CrossRef] [PubMed]
- Spiwok, V.; Kříž, P. Time-Lagged t-Distributed Stochastic Neighbor Embedding (t-SNE) of Molecular Simulation Trajectories. Front. Mol. Biosci. 2020, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Vigil-Vásquez, C.; Schüller, A. De Novo Prediction of Drug Targets and Candidates by Chemical Similarity-Guided Network-Based Inference. Int. J. Mol. Sci. 2022, 23, 9666. [Google Scholar] [CrossRef]
- Yi, J.; Lee, S.; Lim, S.; Cho, C.; Piao, Y.; Yeo, M.; Kim, D.; Kim, S.; Lee, S. Exploring chemical space for lead identification by propagating on chemical similarity network. Comput. Struct. Biotechnol. J. 2023, 21, 4187–4195. [Google Scholar] [CrossRef]
- Ruf, A.; Danger, G. Network Analysis Reveals Spatial Clustering and Annotation of Complex Chemical Spaces: Application to Astrochemistry. Anal. Chem. 2022, 94, 14135–14142. [Google Scholar] [CrossRef]
- Sun, S.; Liu, H.; Guo, Z.; Guan, Q.; Wang, Y.; Wang, J.; Qi, Y.; Yan, Y.; Wang, Y.; Wen, J.; et al. Development and validation of a short-form suboptimal health status questionnaire. EPMA J. 2023, 14, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Wei, J. The adoption of repeated measurement of variance analysis and Shapiro-Wilk test. Front. Med. 2022, 16, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.P.; Malinovsky, Y. Confidence intervals of the Mann-Whitney parameter that are compatible with the Wilcoxon-Mann-Whitney test. Stat. Med. 2018, 37, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Hoijtink, H.; Moerbeek, M. Sample-size determination for the Bayesian t test and Welch’s test using the approximate adjusted fractional Bayes factor. Behav. Res. Methods 2021, 53, 139–152. [Google Scholar] [CrossRef]
- Hermsen, M.; de Bel, T.; den Boer, M.; Steenbergen, E.J.; Kers, J.; Florquin, S.; Roelofs, J.; Stegall, M.D.; Alexander, M.P.; Smith, B.H.; et al. Deep Learning-Based Histopathologic Assessment of Kidney Tissue. J. Am. Soc. Nephrol. 2019, 30, 1968–1979. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Zhong, S.; Zhang, W.; Igou, T.; Berger, E.; Reid, E.; Zhao, Y.; Lambeth, D.; Gan, L.; Afolabi, M.A.; et al. Revolutionizing Membrane Design Using Machine Learning-Bayesian Optimization. Environ. Sci. Technol. 2022, 56, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Yang, Y.X.; Li, J.; Fei, Y.; Guo, H.; Sun, Z.; Lu, J.; Xu, X.; Jiang, Q.; Ikegawa, S.; et al. Molecular Classification of Knee Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 725568. [Google Scholar] [CrossRef]
- do Bomfim, M.R.; Araújo, J.S.C.; Macêdo, W.; Santos, C.; Leite, F.H.A. Identification of potential modulator of Anopheles gambiae odorant binding protein 1 by hierarchical virtual screening and molecular dynamics. J. Biomol. Struct. Dyn. 2021, 39, 6031–6043. [Google Scholar] [CrossRef]
- Borges, R.M.; Colby, S.M.; Das, S.; Edison, A.S.; Fiehn, O.; Kind, T.; Lee, J.; Merrill, A.T.; Merz, K.M., Jr.; Metz, T.O.; et al. Quantum Chemistry Calculations for Metabolomics. Chem. Rev. 2021, 121, 5633–5670. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Molecule Name | Closeness Centrality |
|---|---|---|
| 4,5-Dimethylthiazole Group | 4,5-Dimethylthiazole | 0.16 |
| 6-Methyl-3-hepten-2-one Group | 6-Methyl-3-hepten-2-one | 0.07 |
| Hexyl valerate Group | Hexyl valerate | 0.16 |
| 2-Methyl-4-phenyl-2-butanol Group | 2-Methyl-4-phenyl-2-butanol | 0.20 |
| Features | F1 | F2 | F3 |
|---|---|---|---|
| MolWt | 0.97 | −0.15 | 0.19 |
| FpDensityMorgan1 | −0.41 | 0.85 | 0.06 |
| FpDensityMorgan2 | −0.23 | 0.97 | −0.03 |
| FpDensityMorgan3 | 0.06 | 0.95 | −0.11 |
| HeavyAtomMolWt | 0.96 | −0.14 | 0.23 |
| NumValenceElectrons | 0.97 | −0.16 | 0.16 |
| TPSA | 0.21 | −0.04 | 0.98 |
| pyLabuteASA | 0.98 | −0.15 | 0.11 |
| PSA | 0.21 | −0.04 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Qian, Z.; He, Y.; Liu, M.; Li, W.; Han, W. Mlp4green: A Binary Classification Approach Specifically for Green Odor. Int. J. Mol. Sci. 2024, 25, 3515. https://doi.org/10.3390/ijms25063515
Yang J, Qian Z, He Y, Liu M, Li W, Han W. Mlp4green: A Binary Classification Approach Specifically for Green Odor. International Journal of Molecular Sciences. 2024; 25(6):3515. https://doi.org/10.3390/ijms25063515
Chicago/Turabian StyleYang, Jiuliang, Zhiming Qian, Yi He, Minghao Liu, Wannan Li, and Weiwei Han. 2024. "Mlp4green: A Binary Classification Approach Specifically for Green Odor" International Journal of Molecular Sciences 25, no. 6: 3515. https://doi.org/10.3390/ijms25063515