
Microbiota, Tryptophan and Aryl Hydrocarbon Receptors as the Target Triad in Parkinson’s Disease—A Narrative Review

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gut–Brain Axis Correlates Microbiota with Neuroinflammation
2.1. Gut–Neuroinflammation Axis in PD
2.2. Microbiota and Microbiome in PD
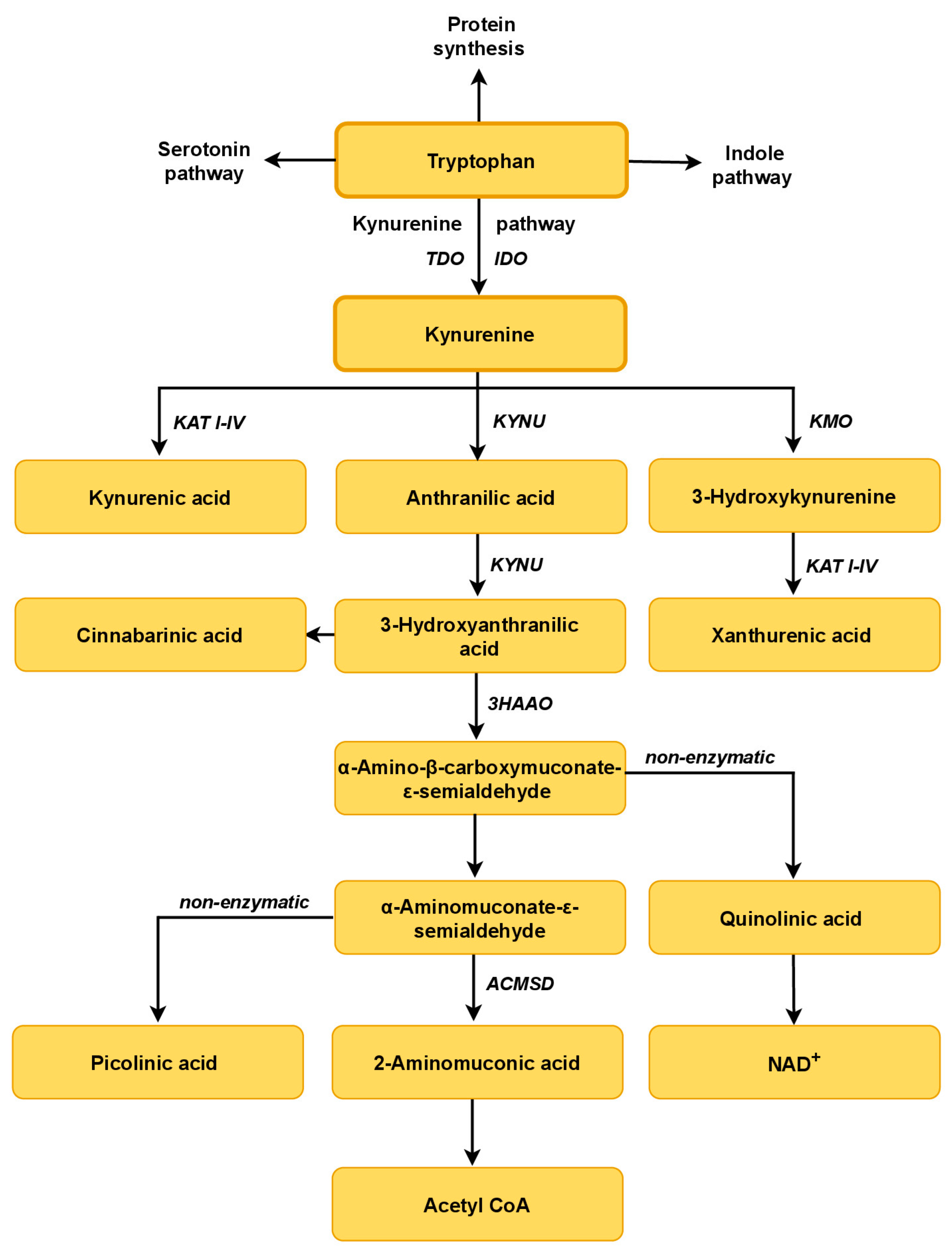
3. Tryptophan Metabolism
3.1. The Tryptophan–Kynurenine Pathway
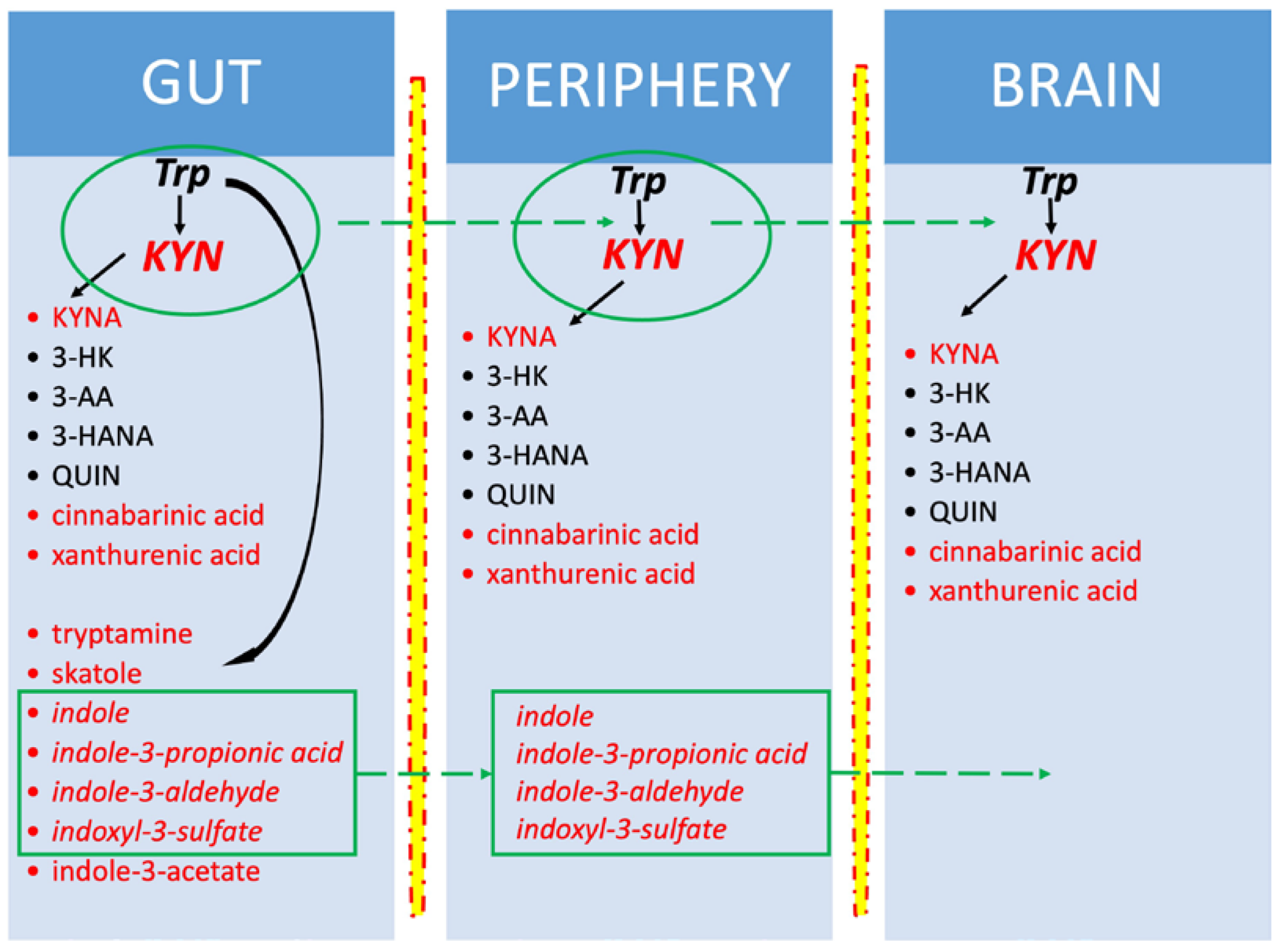
3.2. Trp Metabolism in the Microbiota–Indole Pathway
3.3. Trp-KYN Pathway in PD
4. AhR in the Gut–Brain Axis
4.1. AhR Signaling Pathways
4.2. AhR in PD
5. Dietary Interventions
5.1. Oral Tryptophan and Peripheral Kynurenines
5.2. Oral Tryptophan and Brain Kynurenines
5.3. Antibiotics and Probiotics
5.4. Safety of Dietary Supplementation with Tryptophan
5.5. Fecal Microbiota Transplantation
6. Future Perspectives and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3HAAO | 3-hydroxyanthranilate 3,4-dioxygenase |
| 3-HANA | 3-hydroxyanthranilic acid |
| 3-HAO | 3-hydroxyanthranilic acid 3,4-dioxygenase |
| 3-HK | 3-hydroxykynurenine |
| 6-OHDA | 6-hydroxydopamine |
| AA | anthranilic acid |
| ACMS | α-amino-α-carboxymuconate-ε-semialdehyde |
| ACMSD | α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase |
| AhR | aryl hydrocarbon receptor |
| AIP | protein kinase c-Scr AhR-interacting protein |
| ArAT | aromatic amino acid aminotransferase |
| BBB | Blood–brain barrier |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CYP | cytochrome P450 |
| FMT | fecal microbiota transplantation |
| GPR35 | G-protein-coupled orphan receptor 35 |
| HSP90 | heat shock protein 90 |
| I3S | indoxyl-3-sulfate |
| IAA | indole-3-acetic acid |
| IAAld | indole-3-acetaldehyde |
| IDO | indoleamine 2,3-dioxygenase |
| IE | indole ethanol |
| IL | interleukin |
| ILA | indole-3-lactic acid |
| ILDH | indole lactic acid dehydrogenase |
| IPA | indole-3-propionic acid |
| IS | indoxyl-3-sulfate |
| ITE | 2-(19H-indole-39-carbonyl)-thiazole-4-carboxylic acid methyl ester |
| KAT I–IV | kynurenine aminotransferase I–IV |
| KMO | kynurenine 3-monooxygenase |
| KYN | kynurenine |
| KYNA | kynurenic acid |
| KYNU | kynureninase |
| L-DOPA | levodopa |
| LNAA | large neutral amino acid |
| LNAAT | large neutral amino acid transporter |
| LPHC | low-protein, high-carbohydrate |
| LPS | lipopolysaccharide |
| MHC | major histocompatibility complex |
| MIND | diet Mediterranean-DASH Intervention for Neurodegenerative Delay |
| MPP+ | 1-methyl-4-phenylpyridinium ion |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NAD+ | nicotinamide adenine dinucleotide |
| NF-κB | nuclear factor kappa-light-chain enhancer of activated B cells |
| NMDA | N-methyl-D-aspartate |
| PD | Parkinson’s disease |
| PUFAs | polyunsaturated fatty acids |
| PXR | pregnane X receptor |
| QUIN | quinolinic acid |
| ROS | reactive oxygen species |
| SCFA | short-chain fatty acid |
| SN | substantia nigra |
| Src | Src family kinases |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TDO | tryptophan 2,3-dioxygenase; |
| TLR-4 | Toll-like receptor |
| Trp | tryptophan |
| UbcH7 | ubiquitin-conjugating enzyme H7 |
| XA | xanthurenic acid |
| XRE, DRE | xenobiotic responsive element |
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Genome Wide Association Study. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The Genetics of Parkinson Disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Shen, T.; Yue, Y.; He, T.; Huang, C.; Qu, B.; Lv, W.; Lai, H.-Y. The Association between the Gut Microbiota and Parkinson’s Disease, a Meta-Analysis. Front. Aging Neurosci. 2021, 13, 40. [Google Scholar] [CrossRef]
- Knight, E.; Geetha, T.; Burnett, D.; Babu, J.R. The Role of Diet and Dietary Patterns in Parkinson’s Disease. Nutrients 2022, 14, 4472. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative Stress and Regulated Cell Death in Parkinson’s Disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Jurcau, A.; Andronie-Cioara, F.L.; Nistor-Cseppento, D.C.; Pascalau, N.; Rus, M.; Vasca, E.; Jurcau, M.C. The Involvement of Neuroinflammation in the Onset and Progression of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14582. [Google Scholar] [CrossRef]
- Gries, M.; Christmann, A.; Schulte, S.; Weyland, M.; Rommel, S.; Martin, M.; Baller, M.; Röth, R.; Schmitteckert, S.; Unger, M.; et al. Parkinson Mice Show Functional and Molecular Changes in the Gut Long before Motoric Disease Onset. Mol. Neurodegener. 2021, 16, 34. [Google Scholar] [CrossRef]
- Senevirathne, D.K.L.; Mahboob, A.; Zhai, K.; Paul, P.; Kammen, A.; Lee, D.J.; Yousef, M.S.; Chaari, A. Deep Brain Stimulation beyond the Clinic: Navigating the Future of Parkinson’s and Alzheimer’s Disease Therapy. Cells 2023, 12, 1478. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome Definition Re-Visited: Old Concepts and New Challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Ramos-García, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Gómez Apo, E.; Chávez Macías, L.G.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer’s Patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The Role of Microbiota-Gut-Brain Axis in Neuropsychiatric and Neurological Disorders. Pharmacol. Res. 2021, 172, 105840. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s Disease: Possible Routes by Which Vulnerable Neuronal Types May Be Subject to Neuroinvasion by an Unknown Pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Sun, M.-F.; Shen, Y.-Q. Dysbiosis of Gut Microbiota and Microbial Metabolites in Parkinson’s Disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef]
- Heravi, F.S.; Naseri, K.; Hu, H. Gut Microbiota Composition in Patients with Neurodegenerative Disorders (Parkinson’s and Alzheimer’s) and Healthy Controls: A Systematic Review. Nutrients 2023, 15, 4365. [Google Scholar] [CrossRef]
- Post, Z.; Manfready, R.A.; Keshavarzian, A. Overview of the Gut-Brain Axis: From Gut to Brain and Back Again. Semin. Neurol. 2023, 43, 506–517. [Google Scholar] [CrossRef]
- Hauser, M.; Ray, A. Principles of Riemannian Geometry in Neural Networks. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates, Inc.: Red Hook, NY, USA, 2017; Volume 30. [Google Scholar]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef]
- Savitz, J. The Kynurenine Pathway: A Finger in Every Pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Mu, C.; Farzi, A.; Zhu, W. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Davidson, M.; Rashidi, N.; Nurgali, K.; Apostolopoulos, V. The Role of Tryptophan Metabolites in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 9968. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, C.; Popławski, T.; Chojnacki, J.; Fila, M.; Konrad, P.; Blasiak, J. Tryptophan Intake and Metabolism in Older Adults with Mood Disorders. Nutrients 2020, 12, 3183. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Gilbert, J.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.; Lynch, S.V.; Knight, R. Current Understanding of the Human Microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Yuan, C.; He, Y.; Xie, K.; Feng, L.; Gao, S.; Cai, L. Review of Microbiota Gut Brain Axis and Innate Immunity in Inflammatory and Infective Diseases. Front. Cell. Infect. Microbiol. 2023, 13, 1282431. [Google Scholar] [CrossRef]
- Heiss, C.N.; Olofsson, L.E. The Role of the Gut Microbiota in Development, Function and Disorders of the Central Nervous System and the Enteric Nervous System. J. Neuroendocrinol. 2019, 31, e12684. [Google Scholar] [CrossRef]
- Stolzer, I.; Scherer, E.; Süß, P.; Rothhammer, V.; Winner, B.; Neurath, M.F.; Günther, C. Impact of Microbiome-Brain Communication on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 14925. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Bürmann, J.; Faßbender, K.; Schäfer, K.-H.; Unger, M.M. Fecal Markers of Intestinal Inflammation and Intestinal Permeability Are Elevated in Parkinson’s Disease. Park. Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.-M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L.; et al. Intestinal Infection Triggers Parkinson’s Disease-like Symptoms in Pink1−/− Mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between Microglial Activation and Dopaminergic Neuronal Loss in the Substantia Nigra: A Time Course Study in a 6-Hydroxydopamine Model of Parkinson’s Disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef]
- Chao, Y.-X.; Gulam, M.Y.; Chia, N.S.J.; Feng, L.; Rotzschke, O.; Tan, E.-K. Gut–Brain Axis: Potential Factors Involved in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- García-Revilla, J.; Herrera, A.J.; de Pablos, R.M.; Venero, J.L. Inflammatory Animal Models of Parkinson’s Disease. J. Park. Dis. 2022, 12, S165–S182. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.P.; Carvey, P.M.; Keshavarzian, A.; Shannon, K.M.; Shaikh, M.; Bakay, R.A.E.; Kordower, J.H. Progression of Intestinal Permeability Changes and Alpha-Synuclein Expression in a Mouse Model of Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G.; Dunlop, S.A.; Mastaglia, F.L.; Akkari, P.A.; Koengten, F.; et al. Altered Gut Microbiome in Parkinson’s Disease and the Influence of Lipopolysaccharide in a Human α-Synuclein Over-Expressing Mouse Model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-Synuclein Preformed Fibrils into the Mouse Gastrointestinal Tract Induces Lewy Body-like Aggregates in the Brainstem via the Vagus Nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for Bidirectional and Trans-Synaptic Parasympathetic and Sympathetic Propagation of Alpha-Synuclein in Rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Villarán, R.F.; Espinosa-Oliva, A.M.; Sarmiento, M.; De Pablos, R.M.; Argüelles, S.; Delgado-Cortés, M.J.; Sobrino, V.; Van Rooijen, N.; Venero, J.L.; Herrera, A.J.; et al. Ulcerative Colitis Exacerbates Lipopolysaccharide-Induced Damage to the Nigral Dopaminergic System: Potential Risk Factor in Parkinson`s Disease. J. Neurochem. 2010, 114, 1687–1700. [Google Scholar] [CrossRef]
- McFleder, R.L.; Makhotkina, A.; Groh, J.; Keber, U.; Imdahl, F.; Peña Mosca, J.; Peteranderl, A.; Wu, J.; Tabuchi, S.; Hoffmann, J.; et al. Brain-to-Gut Trafficking of Alpha-Synuclein by CD11c+ Cells in a Mouse Model of Parkinson’s Disease. Nat. Commun. 2023, 14, 7529. [Google Scholar] [CrossRef]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Claudino dos Santos, J.C.; Oliveira, L.F.; Noleto, F.M.; Gusmão, C.T.P.; de Castro Brito, G.A.; de Barros Viana, G.S. Gut-Microbiome-Brain Axis: The Crosstalk between the Vagus Nerve, Alpha-Synuclein and the Brain in Parkinson’s Disease. Neural Regen. Res. 2023, 18, 2611–2614. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, Y.; Si, J.; Pu, M.; Ross, O.A.; McLean, P.J.; Till, L.; Moor, W.; Grover, M.; Kandimalla, K.K.; Margolis, K.G.; et al. Role of Gut Microbiota in Regulating Gastrointestinal Dysfunction and Motor Symptoms in a Mouse Model of Parkinson’s Disease. Gut Microbes 2021, 13, 1866974. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Hashimoto, K. Antibiotic-Induced Microbiome Depletion Protects against MPTP-Induced Dopaminergic Neurotoxicity in the Brain. Aging 2019, 11, 6915–6929. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Langley, M.R.; Palanisamy, B.N.; Dutta, S.; Narayanaswamy, K.; Plummer, P.J.; Sarkar, S.; Ay, M.; Jin, H.; Anantharam, V.; et al. MitoPark Transgenic Mouse Model Recapitulates the Gastrointestinal Dysfunction and Gut-Microbiome Changes of Parkinson’s Disease. Neurotoxicology 2019, 75, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic Stress-Induced Gut Dysfunction Exacerbates Parkinson’s Disease Phenotype and Pathology in a Rotenone-Induced Mouse Model of Parkinson’s Disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut Microbiota Are Related to Parkinson’s Disease and Clinical Phenotype. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Minato, T.; Maeda, T.; Fujisawa, Y.; Tsuji, H.; Nomoto, K.; Ohno, K.; Hirayama, M. Progression of Parkinson’s Disease Is Associated with Gut Dysbiosis: Two-Year Follow-up Study. PLoS ONE 2017, 12, e0187307. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, B.; Zhao, L.-D.; Li, H. The Gut Microbiota: Emerging Evidence in Autoimmune Diseases. Trends Mol. Med. 2020, 26, 862–873. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Papić, E.; Rački, V.; Hero, M.; Tomić, Z.; Starčević-Čižmarević, N.; Kovanda, A.; Kapović, M.; Hauser, G.; Peterlin, B.; Vuletić, V. The Effects of Microbiota Abundance on Symptom Severity in Parkinson’s Disease: A Systematic Review. Front. Aging Neurosci. 2022, 14, 1020172. [Google Scholar] [CrossRef]
- Proano, A.C.; Viteri, J.A.; Orozco, E.N.; Calle, M.A.; Costa, S.C.; Reyes, D.V.; German-Montenegro, M.; Moncayo, D.F.; Tobar, A.C.; Moncayo, J.A. Gut Microbiota and Its Repercussion in Parkinson’s Disease: A Systematic Review in Occidental Patients. Neurol. Int. 2023, 15, 750–763. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Fusco, W.; Lorenzo, M.B.; Cintoni, M.; Porcari, S.; Rinninella, E.; Kaitsas, F.; Lener, E.; Mele, M.C.; Gasbarrini, A.; Collado, M.C.; et al. Short-Chain Fatty-Acid-Producing Bacteria: Key Components of the Human Gut Microbiota. Nutrients 2023, 15, 2211. [Google Scholar] [CrossRef] [PubMed]
- Dănău, A.; Dumitrescu, L.; Lefter, A.; Tulbă, D.; Popescu, B.O. Small Intestinal Bacterial Overgrowth as Potential Therapeutic Target in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 11663. [Google Scholar] [CrossRef]
- Li, X.; Feng, X.; Jiang, Z.; Jiang, Z. Association of Small Intestinal Bacterial Overgrowth with Parkinson’s Disease: A Systematic Review and Meta-Analysis. Gut Pathog. 2021, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.F.; Alonso-Juarez, M. The Link between Gut Dysbiosis and Neuroinflammation in Parkinson’s Disease. Neuroscience 2020, 432, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional Implications of Microbial and Viral Gut Metagenome Changes in Early Stage L-DOPA-Naïve Parkinson’s Disease Patients. Genome Med. 2017, 9, 39. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Gerhardt, S.; Mohajeri, M.H. Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases. Nutrients 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The Nasal and Gut Microbiome in Parkinson’s Disease and Idiopathic Rapid Eye Movement Sleep Behavior Disorder. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Hirayama, M.; Ohno, K. Parkinson’s Disease and Gut Microbiota. Ann. Nutr. Metab. 2021, 77 (Suppl. S2), 28–35. [Google Scholar] [CrossRef]
- Mirzaei, R.; Bouzari, B.; Hosseini-Fard, S.R.; Mazaheri, M.; Ahmadyousefi, Y.; Abdi, M.; Jalalifar, S.; Karimitabar, Z.; Teimoori, A.; Keyvani, H.; et al. Role of Microbiota-Derived Short-Chain Fatty Acids in Nervous System Disorders. Biomed. Pharmacother. 2021, 139, 111661. [Google Scholar] [CrossRef]
- Becker, A.; Faßbender, K.; Oertel, W.H.; Unger, M.M. A Punch in the Gut—Intestinal Inflammation Links Environmental Factors to Neurodegeneration in Parkinson’s Disease. Park. Relat. Disord. 2019, 60, 43–45. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. IJTR 2017, 10, 1178646917691938. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Wisniewska-Jarosiñska, M.; Boznanska-⟪wiçtaszek, P.; Mokwiñska, M.; Stçpieñ, A.; Chojnacki, J. Evaluation of Serotonin and Melatonin Metabolism Indices in Patients with Ulcerative Colitis. Gastroenterol. Pol. Gastroenterol. 2009, 16, 358–362. [Google Scholar]
- Li, D.; Yu, S.; Long, Y.; Shi, A.; Deng, J.; Ma, Y.; Wen, J.; Li, X.; Liu, S.; Zhang, Y.; et al. Tryptophan Metabolism: Mechanism-Oriented Therapy for Neurological and Psychiatric Disorders. Front. Immunol. 2022, 13, 985378. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic Acid in Neurodegenerative Disorders-Unique Neuroprotection or Double-Edged Sword? CNS Neurosci. Ther. 2022, 28, 19–35. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine Pathway Metabolites in Humans: Disease and Healthy States. Int. J. Tryptophan Res. IJTR 2009, 2, IJTR-S2097. [Google Scholar] [CrossRef]
- Marszalek-Grabska, M.; Walczak, K.; Gawel, K.; Wicha-Komsta, K.; Wnorowska, S.; Wnorowski, A.; Turski, W.A. Kynurenine Emerges from the Shadows—Current Knowledge on Its Fate and Function. Pharmacol. Ther. 2021, 225, 107845. [Google Scholar] [CrossRef]
- Han, Q.; Cai, T.; Tagle, D.; Li, J. Thermal Stability, pH Dependence and Inhibition of Four Murine Kynurenine Aminotransferases. BMC Biochem. 2010, 11, 19. [Google Scholar] [CrossRef]
- Reyes Ocampo, J.; Lugo Huitrón, R.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; Pérez de la Cruz, V. Kynurenines with Neuroactive and Redox Properties: Relevance to Aging and Brain Diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Monitoring the Kynurenine System: Concentrations, Ratios or What Else? Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. 2021, 30, 775–778. [Google Scholar] [CrossRef]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An Expanding Range of Targets for Kynurenine Metabolites of Tryptophan. Trends Pharmacol. Sci. 2013, 34, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Copeland, C.S.; Neale, S.A.; Bruno, V.; Battaglia, G.; Salt, T.E.; Nicoletti, F. Cinnabarinic Acid and Xanthurenic Acid: Two Kynurenine Metabolites That Interact with Metabotropic Glutamate Receptors. Neuropharmacology 2017, 112, 365–372. [Google Scholar] [CrossRef]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated Brain 3-Hydroxykynurenine and Quinolinate Levels in Huntington Disease Mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef]
- Luthra, M.; Balasubramanian, D. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid May Act as Endogenous Antioxidants in the Eye Lens. Exp. Eye Res. 1992, 55, 641–643. [Google Scholar] [CrossRef]
- Stone, T.W.; Perkins, M.N. Quinolinic Acid: A Potent Endogenous Excitant at Amino Acid Receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.; Suh, H.-S.; Tarassishin, L.; Cui, Q.L.; Durafourt, B.A.; Choi, N.; Bauman, A.; Cosenza-Nashat, M.; Antel, J.P.; Zhao, M.-L.; et al. The Tryptophan Metabolite 3-Hydroxyanthranilic Acid Plays Anti-Inflammatory and Neuroprotective Roles during Inflammation: Role of Hemeoxygenase-1. Am. J. Pathol. 2011, 179, 1360–1372. [Google Scholar] [CrossRef]
- Mizdrak, J.; Hains, P.G.; Truscott, R.J.W.; Jamie, J.F.; Davies, M.J. Tryptophan-Derived Ultraviolet Filter Compounds Covalently Bound to Lens Proteins Are Photosensitizers of Oxidative Damage. Free Radic. Biol. Med. 2008, 44, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, D.; Tankiewicz, A.; Matys, T.; Buczko, W. Peripheral Distribution of Kynurenine Metabolites and Activity of Kynurenine Pathway Enzymes in Renal Failure. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2003, 54, 175–189. [Google Scholar]
- Sala, A.; Campagnoli, M.; Perani, E.; Romano, A.; Labò, S.; Monzani, E.; Minchiotti, L.; Galliano, M. Human Alpha-1-Microglobulin Is Covalently Bound to Kynurenine-Derived Chromophores. J. Biol. Chem. 2004, 279, 51033–51041. [Google Scholar] [CrossRef]
- Hood, B.D.; Garner, B.; Truscott, R.J. Human Lens Coloration and Aging. Evidence for Crystallin Modification by the Major Ultraviolet Filter, 3-Hydroxy-Kynurenine O-Beta-D-Glucoside. J. Biol. Chem. 1999, 274, 32547–32550. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C.; et al. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Generate Hydrogen Peroxide and Promote Alpha-Crystallin Cross-Linking by Metal Ion Reduction. Biochemistry 2000, 39, 7266–7275. [Google Scholar] [CrossRef]
- Parker, N.R.; Jamie, J.F.; Davies, M.J.; Truscott, R.J.W. Protein-Bound Kynurenine Is a Photosensitizer of Oxidative Damage. Free Radic. Biol. Med. 2004, 37, 1479–1489. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis during Early Life Regulates the Hippocampal Serotonergic System in a Sex-Dependent Manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Cross, P.J.; Dobson, R.C.J.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A Three-Ring Circus: Metabolism of the Three Proteogenic Aromatic Amino Acids and Their Role in the Health of Plants and Animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef]
- Lee, S.-J.; Lim, H.-S.; Masliah, E.; Lee, H.-J. Protein Aggregate Spreading in Neurodegenerative Diseases: Problems and Perspectives. Neurosci. Res. 2011, 70, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Tennoune, N.; Andriamihaja, M.; Blachier, F. Production of Indole and Indole-Related Compounds by the Intestinal Microbiota and Consequences for the Host: The Good, the Bad, and the Ugly. Microorganisms 2022, 10, 930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, W.-J.; Quan, W.; Qiao, C.-M.; Cui, C.; Hong, H.; Shi, Y.; Niu, G.-Y.; Zhao, L.-P.; Shen, Y.-Q. Dynamic Changes of Activated AHR in Microglia and Astrocytes in the Substantia Nigra-Striatum System in an MPTP-Induced Parkinson’s Disease Mouse Model. Brain Res. Bull. 2021, 176, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 Impacts Colitis by Altering Gut Microbiota Metabolism of Tryptophan into Aryl Hydrocarbon Receptor Ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A Gut Bacterial Pathway Metabolizes Aromatic Amino Acids into Nine Circulating Metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major Phenylpropanoid-Derived Metabolites in the Human Gut Can Arise from Microbial Fermentation of Protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; He, H.; Peng, M.; Zeng, M.; Sun, H. The Role of the Indoles in Microbiota-Gut-Brain Axis and Potential Therapeutic Targets: A Focus on Human Neurological and Neuropsychiatric Diseases. Neuropharmacology 2023, 239, 109690. [Google Scholar] [CrossRef]
- Török, N.; Maszlag-Török, R.; Molnár, K.; Szolnoki, Z.; Somogyvári, F.; Boda, K.; Tanaka, M.; Klivényi, P.; Vécsei, L. Single Nucleotide Polymorphisms of Indoleamine 2,3-Dioxygenase Influenced the Age Onset of Parkinson’s Disease. Front. Biosci.-Landmark 2022, 27, 265. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. S1), 318–324. [Google Scholar] [CrossRef] [PubMed]
- Bryleva, E.Y.; Keaton, S.A.; Grit, J.; Madaj, Z.; Sauro-Nagendra, A.; Smart, L.; Halstead, S.; Achtyes, E.; Brundin, L. The Acute-Phase Mediator Serum Amyloid A Is Associated with Symptoms of Depression and Fatigue. Acta Psychiatr. Scand. 2017, 135, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed]
- Luchowski, P.; Luchowska, E.; Turski, W.A.; Urbanska, E.M. 1-Methyl-4-Phenylpyridinium and 3-Nitropropionic Acid Diminish Cortical Synthesis of Kynurenic Acid via Interference with Kynurenine Aminotransferases in Rats. Neurosci. Lett. 2002, 330, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Knyihár-Csillik, E.; Chadaide, Z.; Mihály, A.; Krisztin-Péva, B.; Fenyo, R.; Vécsei, L. Effect of 6-Hydroxydopamine Treatment on Kynurenine Aminotransferase-I (KAT-I) Immunoreactivity of Neurons and Glial Cells in the Rat Substantia Nigra. Acta Neuropathol. 2006, 112, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Brotchie, J.M.; Mitchell, I.J.; Sambrook, M.A.; Crossman, A.R. Alleviation of Parkinsonism by Antagonism of Excitatory Amino Acid Transmission in the Medial Segment of the Globus Pallidus in Rat and Primate. Mov. Disord. Off. J. Mov. Disord. Soc. 1991, 6, 133–138. [Google Scholar] [CrossRef]
- Butler, E.G.; Bourke, D.W.; Finkelstein, D.I.; Horne, M.K. The Effects of Reversible Inactivation of the Subthalamo-Pallidal Pathway on the Behaviour of Naive and Hemiparkinsonian Monkeys. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 1997, 4, 218–227. [Google Scholar] [CrossRef]
- Silva-Adaya, D.; Pérez-De La Cruz, V.; Villeda-Hernández, J.; Carrillo-Mora, P.; González-Herrera, I.G.; García, E.; Colín-Barenque, L.; Pedraza-Chaverrí, J.; Santamaría, A. Protective Effect of L-Kynurenine and Probenecid on 6-Hydroxydopamine-Induced Striatal Toxicity in Rats: Implications of Modulating Kynurenate as a Protective Strategy. Neurotoxicol. Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Izzo, E.; Schwarcz, R.; Bédard, P.J. Effect of Kynurenine 3-Hydroxylase Inhibition on the Dyskinetic and Antiparkinsonian Responses to Levodopa in Parkinsonian Monkeys. Mov. Disord. Off. J. Mov. Disord. Soc. 2005, 20, 792–802. [Google Scholar] [CrossRef]
- Szabó, N.; Kincses, Z.T.; Toldi, J.; Vécsei, L. Altered Tryptophan Metabolism in Parkinson’s Disease: A Possible Novel Therapeutic Approach. J. Neurol. Sci. 2011, 310, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Knyihár-Csillik, E.; Csillik, B.; Pákáski, M.; Krisztin-Péva, B.; Dobó, E.; Okuno, E.; Vécsei, L. Decreased Expression of Kynurenine Aminotransferase-I (KAT-I) in the Substantia Nigra of Mice after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Treatment. Neuroscience 2004, 126, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huan, F.; Wang, J.; Xie, X.; Yu, G.; Wang, X.; Jiang, L.; Gao, R.; Xiao, H.; Ding, H.; et al. 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Induced Parkinson’s Disease in Mouse: Potential Association between Neurotransmitter Disturbance and Gut Microbiota Dysbiosis. ACS Chem. Neurosci. 2020, 11, 3366–3376. [Google Scholar] [CrossRef]
- Boros, F.A.; Vécsei, L. Tryptophan 2,3-Dioxygenase, a Novel Therapeutic Target for Parkinson’s Disease. Expert Opin. Ther. Targets 2021, 25, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, G.C.; Targa, A.D.S.; Soares-Lima, S.C.; dos Santos, P.I.; Rodrigues, L.S.; Macedo, D.A.; Ribeiro Pinto, L.F.; Lima, M.M.S.; Boldt, A.B.W. Folic Acid and Vitamin B12 Prevent Deleterious Effects of Rotenone on Object Novelty Recognition Memory and Kynu Expression in an Animal Model of Parkinson’s Disease. Genes 2022, 13, 2397. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.; van der Hart, M.; Roeser, J.; Summergrad, P. Peripheral Tryptophan—Kynurenine Metabolism Associated with Metabolic Syndrome Is Different in Parkinson’s and Alzheimer’s Diseases. Endocrinol. Diabetes Metab. J. 2017, 1, 1–5. Available online: https://researchopenworld.com/wp-content/uploads/2017/11/EDMJ-2017-113-Gregory-F-Oxenkrug-USA.pdf (accessed on 7 October 2023).
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine Metabolism in Plasma and in Red Blood Cells in Parkinson’s Disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Cheng, M.-L.; Tang, H.-Y.; Huang, C.-Y.; Wu, Y.-R.; Chen, C.-M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef]
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X.; et al. Tryptophan Metabolites Are Associated with Symptoms and Nigral Pathology in Parkinson’s Disease. Mov. Disord. 2020, 35, 2028–2037. [Google Scholar] [CrossRef]
- Fathi, M.; Taghizadeh, F.; Mojtahedi, H.; Zargar Balaye Jame, S.; Markazi Moghaddam, N. The Effects of Alzheimer’s and Parkinson’s Disease on 28-Day Mortality of COVID-19. Rev. Neurol. 2022, 178, 129–136. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine Pathway Abnormalities in Parkinson’s Disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef]
- Nordengen, K.; Morland, C. From Synaptic Physiology to Synaptic Pathology: The Enigma of α-Synuclein. Int. J. Mol. Sci. 2024, 25, 986. [Google Scholar] [CrossRef]
- Saramowicz, K.; Siwecka, N.; Galita, G.; Kucharska-Lusina, A.; Rozpędek-Kamińska, W.; Majsterek, I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 360. [Google Scholar] [CrossRef] [PubMed]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic Acid Amyloid-like Fibrillar Assemblies Seed α-Synuclein Aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef] [PubMed]
- Capucciati, A.; Galliano, M.; Bubacco, L.; Zecca, L.; Casella, L.; Monzani, E.; Nicolis, S. Neuronal Proteins as Targets of 3-Hydroxykynurenine: Implications in Neurodegenerative Diseases. ACS Chem. Neurosci. 2019, 10, 3731–3739. [Google Scholar] [CrossRef]
- Lukić, I.; Ivković, S.; Mitić, M.; Adžić, M. Tryptophan Metabolites in Depression: Modulation by Gut Microbiota. Front. Behav. Neurosci. 2022, 16, 987697. [Google Scholar] [CrossRef]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. The Aryl Hydrocarbon Receptor and the Gut-Brain Axis. Cell. Mol. Immunol. 2021, 18, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jin, U.-H.; Karki, K.; Jayaraman, A.; Allred, C.; Michelhaugh, S.K.; Mittal, S.; Chapkin, R.S.; Safe, S. Dopamine Is an Aryl Hydrocarbon Receptor Agonist. Biochem. J. 2020, 477, 3899–3910. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Tasheva, S.; Soler, R.M. NF-κB Signaling Pathways: Role in Nervous System Physiology and Pathology. Neuroscientist 2013, 19, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Porrini, V.; Bellucci, A.; Benarese, M.; Branca, C.; Parrella, E.; Spano, P.F.; Pizzi, M. NF-κB in Innate Neuroprotection and Age-Related Neurodegenerative Diseases. Front. Neurol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef]
- González-Barbosa, E.; Mejía-García, A.; Bautista, E.; Gonzalez, F.J.; Segovia, J.; Elizondo, G. TCDD Induces UbcH7 Expression and Synphilin-1 Protein Degradation in the Mouse Ventral Midbrain. J. Biochem. Mol. Toxicol. 2017, 31, e21947. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I Interferons and Microbial Metabolites of Tryptophan Modulate Astrocyte Activity and Central Nervous System Inflammation via the Aryl Hydrocarbon Receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Lin, C.-H.; Hsu, P.-C.; Sun, Y.-Y.; Huang, Y.-J.; Zhuo, J.-H.; Wang, C.-Y.; Gan, Y.-L.; Hung, C.-C.; Kuan, C.-Y.; et al. Aryl Hydrocarbon Receptor Mediates Both Proinflammatory and Anti-Inflammatory Effects in Lipopolysaccharide-Activated Microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef]
- Ma, N.; He, T.; Johnston, L.J.; Ma, X. Host–Microbiome Interactions: The Aryl Hydrocarbon Receptor as a Critical Node in Tryptophan Metabolites to Brain Signaling. Gut Microbes 2020, 11, 1203–1219. [Google Scholar] [CrossRef]
- Kuter, K.Z.; Śmiałowska, M.; Ossowska, K. The Influence of Preconditioning with Low Dose of LPS on Paraquat-Induced Neurotoxicity, Microglia Activation and Expression of α-Synuclein and Synphilin-1 in the Dopaminergic System. Pharmacol. Rep. 2022, 74, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Oliveros, A.; Dominguez-Baleón, C.; Sánchez-Díaz, I.; Zambrano-Tipan, D.; Hernández-Vargas, R.; Campusano, J.M.; Narváez-Padilla, V.; Reynaud, E. Parkinsonian Phenotypes Induced by Synphilin-1 Expression Are Differentially Contributed by Serotonergic and Dopaminergic Circuits and Suppressed by Nicotine Treatment. PLoS ONE 2023, 18, e0282348. [Google Scholar] [CrossRef] [PubMed]
- Miranda, B.R.D.; Miller, J.A.; Hansen, R.J.; Lunghofer, P.J.; Safe, S.; Gustafson, D.L.; Colagiovanni, D.; Tjalkens, R.B. Neuroprotective Efficacy and Pharmacokinetic Behavior of Novel Anti-Inflammatory Para-Phenyl Substituted Diindolylmethanes in a Mouse Model of Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2013, 345, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Modoux, M.; Rolhion, N.; Mani, S.; Sokol, H. Tryptophan Metabolism as a Pharmacological Target. Trends Pharmacol. Sci. 2021, 42, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. IJTR 2009, 2, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Sadok, I.; Jędruchniewicz, K. Dietary Kynurenine Pathway Metabolites—Source, Fate, and Chromatographic Determinations. Int. J. Mol. Sci. 2023, 24, 16304. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Tryptophan Availability for Kynurenine Pathway Metabolism across the Life Span: Control Mechanisms and Focus on Aging, Exercise, Diet and Nutritional Supplements. Neuropharmacology 2017, 112, 248–263. [Google Scholar] [CrossRef]
- Hu, D.; Liu, J.; Yu, W.; Li, C.; Huang, L.; Mao, W.; Lu, Z. Tryptophan Intake, Not Always the More the Better. Front. Nutr. 2023, 10, 1140054. [Google Scholar] [CrossRef]
- Hiratsuka, C.; Fukuwatari, T.; Shibata, K. Fate of Dietary Tryptophan in Young Japanese Women. Int. J. Tryptophan Res. IJTR 2012, 5, 33–47. [Google Scholar] [CrossRef]
- Chojnacki, C.; Gąsiorowska, A.; Popławski, T.; Konrad, P.; Chojnacki, M.; Fila, M.; Blasiak, J. Beneficial Effect of Increased Tryptophan Intake on Its Metabolism and Mental State of the Elderly. Nutrients 2023, 15, 847. [Google Scholar] [CrossRef]
- Sathyasaikumar, K.V.; Notarangelo, F.M.; Kelly, D.L.; Rowland, L.M.; Hare, S.M.; Chen, S.; Mo, C.; Buchanan, R.W.; Schwarcz, R. Tryptophan Challenge in Healthy Controls and People with Schizophrenia: Acute Effects on Plasma Levels of Kynurenine, Kynurenic Acid and 5-Hydroxyindoleacetic Acid. Pharmaceuticals 2022, 15, 1003. [Google Scholar] [CrossRef]
- Hare, S.M.; Adhikari, B.M.; Mo, C.; Chen, S.; Wijtenburg, S.A.; Seneviratne, C.; Kane-Gerard, S.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Schwarcz, R.; et al. Tryptophan Challenge in Individuals with Schizophrenia and Healthy Controls: Acute Effects on Circulating Kynurenine and Kynurenic Acid, Cognition and Cerebral Blood Flow. Neuropsychopharmacology 2023, 48, 1594–1601. [Google Scholar] [CrossRef]
- Karlsson, T.; Strand, E.; Dierkes, J.; Drevon, C.A.; Øyen, J.; Midttun, Ø.; Ueland, P.M.; Gudbrandsen, O.A.; Pedersen, E.R.; Nygård, O. Associations between Intake of Fish and N-3 Long-Chain Polyunsaturated Fatty Acids and Plasma Metabolites Related to the Kynurenine Pathway in Patients with Coronary Artery Disease. Eur. J. Nutr. 2017, 56, 261–272. [Google Scholar] [CrossRef]
- Okuno, A.; Fukuwatari, T.; Shibata, K. High Tryptophan Diet Reduces Extracellular Dopamine Release via Kynurenic Acid Production in Rat Striatum. J. Neurochem. 2011, 118, 796–805. [Google Scholar] [CrossRef]
- Melancon, M.O.; Lorrain, D.; Dionne, I.J. Exercise Increases Tryptophan Availability to the Brain in Older Men Age 57–70 Years. Med. Sci. Sports Exerc. 2012, 44, 881–887. [Google Scholar] [CrossRef]
- Campbell, S.C.; Wisniewski, P.J.; Noji, M.; McGuinness, L.R.; Häggblom, M.M.; Lightfoot, S.A.; Joseph, L.B.; Kerkhof, L.J. The Effect of Diet and Exercise on Intestinal Integrity and Microbial Diversity in Mice. PLoS ONE 2016, 11, e0150502. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Pérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; González-Soltero, R.; Larrosa, M. Gut Microbiota Modification: Another Piece in the Puzzle of the Benefits of Physical Exercise in Health? Front. Physiol. 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid. Med. Cell. Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Berg Miller, M.E.; Pence, B.D.; Whitlock, K.; Nehra, V.; Gaskins, H.R.; White, B.A.; Fryer, J.D.; Woods, J.A. Voluntary and Forced Exercise Differentially Alters the Gut Microbiome in C57BL/6J Mice. J. Appl. Physiol. 2015, 118, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Morris, L.S.; Marchesi, J.R. The Gut Microbiome: The Role of a Virtual Organ in the Endocrinology of the Host. J. Endocrinol. 2013, 218, R37–R47. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.; Mermier, C.; Zuhl, M. Exercise Influence on the Microbiome–Gut–Brain Axis. Gut Microbes 2019, 10, 555–568. [Google Scholar] [CrossRef]
- Allen, J.M.; Mailing, L.J.; Cohrs, J.; Salmonson, C.; Fryer, J.D.; Nehra, V.; Hale, V.L.; Kashyap, P.; White, B.A.; Woods, J.A. Exercise Training-Induced Modification of the Gut Microbiota Persists after Microbiota Colonization and Attenuates the Response to Chemically-Induced Colitis in Gnotobiotic Mice. Gut Microbes 2018, 9, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Batacan, R.B.; Fenning, A.S.; Dalbo, V.J.; Scanlan, A.T.; Duncan, M.J.; Moore, R.J.; Stanley, D. A Gut Reaction: The Combined Influence of Exercise and Diet on Gastrointestinal Microbiota in Rats. J. Appl. Microbiol. 2017, 122, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Goetz, L.; Pervaiz, N.; Packer, N.; Guan, J. Freewheel Training Decreases Pro- and Increases Anti-Inflammatory Cytokine Expression in Mouse Intestinal Lymphocytes. Brain. Behav. Immun. 2010, 24, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Packer, N.; Hoffman-Goetz, L. Apoptotic and Inflammatory Cytokine Protein Expression in Intestinal Lymphocytes after Acute Treadmill Exercise in Young and Old Mice. J. Sports Med. Phys. Fitness 2012, 52, 202–211. [Google Scholar]
- Yusufu, I.; Ding, K.; Smith, K.; Wankhade, U.D.; Sahay, B.; Patterson, G.T.; Pacholczyk, R.; Adusumilli, S.; Hamrick, M.W.; Hill, W.D.; et al. A Tryptophan-Deficient Diet Induces Gut Microbiota Dysbiosis and Increases Systemic Inflammation in Aged Mice. Int. J. Mol. Sci. 2021, 22, 5005. [Google Scholar] [CrossRef]
- Lee, C.; Hong, S.N.; Paik, N.Y.; Kim, T.J.; Kim, E.R.; Chang, D.K.; Kim, Y.-H. CD1d Modulates Colonic Inflammation in NOD2−/− Mice by Altering the Intestinal Microbial Composition Comprising Acetatifactor Muris. J. Crohns Colitis 2019, 13, 1081–1091. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut Microbiota Depletion from Early Adolescence in Mice: Implications for Brain and Behaviour. Brain. Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef]
- Gao, K.; Pi, Y.; Peng, Y.; Mu, C.-L.; Zhu, W.-Y. Time-Course Responses of Ileal and Fecal Microbiota and Metabolite Profiles to Antibiotics in Cannulated Pigs. Appl. Microbiol. Biotechnol. 2018, 102, 2289–2299. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, M.; Yang, Y.; Mu, C.; Su, Y.; Zhu, W. Differential Effect of Early Antibiotic Intervention on Bacterial Fermentation Patterns and Mucosal Gene Expression in the Colon of Pigs under Diets with Different Protein Levels. Appl. Microbiol. Biotechnol. 2017, 101, 2493–2505. [Google Scholar] [CrossRef]
- Jiang, P.; Trimigno, A.; Stanstrup, J.; Khakimov, B.; Viereck, N.; Engelsen, S.B.; Sangild, P.T.; Dragsted, L.O. Antibiotic Treatment Preventing Necrotising Enterocolitis Alters Urinary and Plasma Metabolomes in Preterm Pigs. J. Proteome Res. 2017, 16, 3547–3557. [Google Scholar] [CrossRef]
- Mu, C.; Yang, Y.; Yu, K.; Yu, M.; Zhang, C.; Su, Y.; Zhu, W. Alteration of Metabolomic Markers of Amino-Acid Metabolism in Piglets with in-Feed Antibiotics. Amino Acids 2017, 49, 771–781. [Google Scholar] [CrossRef]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Bienenstock, J.; Dinan, T.G. The Probiotic Bifidobacteria Infantis: An Assessment of Potential Antidepressant Properties in the Rat. J. Psychiatr. Res. 2008, 43, 164–174. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus Reuteri Induces Gut Intraepithelial CD4+CD8αα+ T Cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef]
- Rudzki, L.; Ostrowska, L.; Pawlak, D.; Małus, A.; Pawlak, K.; Waszkiewicz, N.; Szulc, A. Probiotic Lactobacillus Plantarum 299v Decreases Kynurenine Concentration and Improves Cognitive Functions in Patients with Major Depression: A Double-Blind, Randomized, Placebo Controlled Study. Psychoneuroendocrinology 2019, 100, 213–222. [Google Scholar] [CrossRef]
- Chu, C.; Li, T.; Yu, L.; Li, Y.; Li, M.; Guo, M.; Zhao, J.; Zhai, Q.; Tian, F.; Chen, W. A Low-Protein, High-Carbohydrate Diet Exerts a Neuroprotective Effect on Mice with 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinson’s Disease by Regulating the Microbiota-Metabolite-Brain Axis and Fibroblast Growth Factor 21. J. Agric. Food Chem. 2023, 71, 8877–8893. [Google Scholar] [CrossRef] [PubMed]
- Cynober, L.; Bier, D.M.; Kadowaki, M.; Morris, S.M.; Elango, R.; Smriga, M. Proposals for Upper Limits of Safe Intake for Arginine and Tryptophan in Young Adults and an Upper Limit of Safe Intake for Leucine in the Elderly. J. Nutr. 2016, 146, 2652S–2654S. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl Sulfate—The Uremic Toxin Linking Hemostatic System Disturbances with the Prevalence of Cardiovascular Disease in Patients with Chronic Kidney Disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef]
- D’Haens, G.R.; Jobin, C. Fecal Microbial Transplantation for Diseases Beyond Recurrent Clostridium Difficile Infection. Gastroenterology 2019, 157, 624–636. [Google Scholar] [CrossRef]
- Dempsey, E.; Corr, S.C. Lactobacillus spp. for Gastrointestinal Health: Current and Future Perspectives. Front. Immunol. 2022, 13, 840245. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-D.; Jenq, R.R.; Ajami, N.J.; Petrosino, J.F.; Alexander, A.A.; Ke, S.; Iqbal, T.; DuPont, A.W.; Muldrew, K.; Shi, Y.; et al. Safety and Preliminary Efficacy of Orally Administered Lyophilized Fecal Microbiota Product Compared with Frozen Product given by Enema for Recurrent Clostridium Difficile Infection: A Randomized Clinical Trial. PLoS ONE 2018, 13, e0205064. [Google Scholar] [CrossRef] [PubMed]
- Cherbut, C.; Aubé, A.C.; Blottière, H.M.; Galmiche, J.P. Effects of Short-Chain Fatty Acids on Gastrointestinal Motility. Scand. J. Gastroenterol. Suppl. 1997, 222, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mocanu, V.; Cai, C.; Dang, J.; Slater, L.; Deehan, E.C.; Walter, J.; Madsen, K.L. Impact of Fecal Microbiota Transplantation on Obesity and Metabolic Syndrome-A Systematic Review. Nutrients 2019, 11, 2291. [Google Scholar] [CrossRef] [PubMed]
- Luqman, A.; Hassan, A.; Ullah, M.; Naseem, S.; Ullah, M.; Zhang, L.; Din, A.U.; Ullah, K.; Ahmad, W.; Wang, G. Role of the Intestinal Microbiome and Its Therapeutic Intervention in Cardiovascular Disorder. Front. Immunol. 2024, 15, 1321395. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, W.; Li, Y. The Interplay between Microbiota and Brain-Gut Axis in Epilepsy Treatment. Front. Pharmacol. 2024, 15, 1276551. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Wang, C.; Bai, J.; Song, J.; Zhang, Y.; Chen, H.; Suo, H. Gut Microbiome-Based Therapies for Alleviating Cognitive Impairment: State of the Field, Limitations, and Future Perspectives. Food Funct. 2024, 15, 1116–1134. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-F.; Zhu, Y.-L.; Zhou, Z.-L.; Jia, X.-B.; Xu, Y.-D.; Yang, Q.; Cui, C.; Shen, Y.-Q. Neuroprotective Effects of Fecal Microbiota Transplantation on MPTP-Induced Parkinson’s Disease Mice: Gut Microbiota, Glial Reaction and TLR4/TNF-α Signaling Pathway. Brain. Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Zhong, Z.; Chen, W.; Gao, H.; Che, N.; Xu, M.; Yang, L.; Zhang, Y.; Ye, M. Fecal Microbiota Transplantation Exerts a Protective Role in MPTP-Induced Parkinson’s Disease via the TLR4/PI3K/AKT/NF-κB Pathway Stimulated by α-Synuclein. Neurochem. Res. 2021, 46, 3050–3058. [Google Scholar] [CrossRef]
- Xue, L.-J.; Yang, X.-Z.; Tong, Q.; Shen, P.; Ma, S.-J.; Wu, S.-N.; Zheng, J.-L.; Wang, H.-G. Fecal Microbiota Transplantation Therapy for Parkinson’s Disease: A Preliminary Study. Medicine 2020, 99, e22035. [Google Scholar] [CrossRef]
- Kuai, X.-Y.; Yao, X.-H.; Xu, L.-J.; Zhou, Y.-Q.; Zhang, L.-P.; Liu, Y.; Pei, S.-F.; Zhou, C.-L. Evaluation of Fecal Microbiota Transplantation in Parkinson’s Disease Patients with Constipation. Microb. Cell Fact. 2021, 20, 98. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwaniak, P.; Owe-Larsson, M.; Urbańska, E.M. Microbiota, Tryptophan and Aryl Hydrocarbon Receptors as the Target Triad in Parkinson’s Disease—A Narrative Review. Int. J. Mol. Sci. 2024, 25, 2915. https://doi.org/10.3390/ijms25052915
Iwaniak P, Owe-Larsson M, Urbańska EM. Microbiota, Tryptophan and Aryl Hydrocarbon Receptors as the Target Triad in Parkinson’s Disease—A Narrative Review. International Journal of Molecular Sciences. 2024; 25(5):2915. https://doi.org/10.3390/ijms25052915
Chicago/Turabian StyleIwaniak, Paulina, Maja Owe-Larsson, and Ewa M. Urbańska. 2024. "Microbiota, Tryptophan and Aryl Hydrocarbon Receptors as the Target Triad in Parkinson’s Disease—A Narrative Review" International Journal of Molecular Sciences 25, no. 5: 2915. https://doi.org/10.3390/ijms25052915