
XB2Bi2 (X = Si, Ge, Sn, Pb): Penta-Atomic Planar Tetracoordinate Si/Ge/Sn/Pb Clusters with 20 Valence Electrons

Abstract
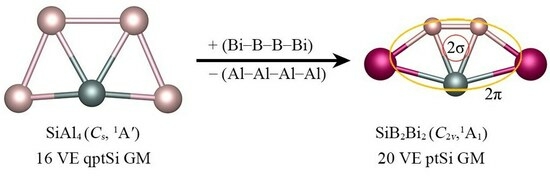
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Structure and Stabilities
2.2. CMOs and AdNDP Analyses
2.3. 2π + 2σ Aromaticity
2.4. Simulated IR Spectra
3. Methods and Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van’t Hoff, J.H. Sur Les Formules De Structure Dans L’espace. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le Bel, J.A. Sur Les Relations Qui Existent Entre Les Formules Atomiques Des Corps Organiques Et Le Pouvoir Rotatoire De Leurs Dissolutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Monkhorst, H.J. Activation Energy for Interconversion of Enantiomers Containing an Asymmetric Carbon Atom without Breaking Bonds. Chem. Commun. 1968, 18, 1111–1112. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.v.R.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Sorger, K.; Schleyer, P.v.R. Planar and Inherently Non-tetrahedral Tetracoordinate Carbon: A Status Report. J. Mol. Struct. 1995, 338, 317–346. [Google Scholar] [CrossRef]
- Erker, G.; Röttger, D. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. Engl. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Erker, G. Using Bent Metallocenes for Stabilizing Unusual Coordination Geometries at Carbon. Chem. Soc. Rev. 1999, 28, 307–314. [Google Scholar] [CrossRef]
- Siebert, W.; Gunale, A. Compounds Containing a Planar-Tetracoordinate Carbon Atom as Analogues of Planar Methane. Chem. Soc. Rev. 1999, 28, 367–371. [Google Scholar] [CrossRef]
- Keese, R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Vela, A.; Heine, T. Recent Advances in Planar Tetracoordinate Carbon Chemistry. J. Comput. Chem. 2007, 28, 362–372. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.F.; Wang, Z.X.; Schleyer, P.v.R. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar Pentacoordinate Carbons. Nat. Rev. Chem. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. Structure and Bonding in Planar Hypercoordinate Carbon Compounds. Chemistry 2022, 4, 1723–1756. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Chen, Z. Planar Hypercoordinate Motifs in Two-Dimensional Materials. Acc. Chem. Res. 2020, 53, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and Ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.D.; Bodyrev, A.I. Pentaatomic Tetracoordinate Plannar Carbon CAl42–: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon–Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Zhang, C.J.; Dai, W.S.; Xu, H.G.; Xu, X.L.; Zheng, W.J. Structural Evolution of Carbon-Doped Aluminum Clusters AlnC− (n = 6–15): Anion Photoelectron Spectroscopy and Theoretical Calculations. J. Phys. Chem. 2022, 126, 5621–5632. [Google Scholar] [CrossRef]
- Zhang, C.J.; Wang, P.; Xu, X.L.; Xu, H.G.; Zheng, W.J. Photoelectron Spectroscopy and Theoretical Study of AlnC5−/0 (n = 1–5) clusters: Structural Evolution, Relative Stability of Star-Like Cluster, and Planar Tetracoordinate Carbon Structure. Phys. Chem. Chem. Phys. 2021, 23, 1967–1975. [Google Scholar] [CrossRef]
- Bai, L.X.; Barroso, J.; Orozco-Ic, M.; Ortiz-Chi, F.; Guo, J.C.; Merino, G. CAl11−: A Molecular Rotor with a Quasi-Planar Tetracoordinate Carbon. Chem. Commun. 2023, 59, 4966–4969. [Google Scholar] [CrossRef]
- Pei, Y.; An, W.; Ito, K.; Schleyer, P.v.R.; Zeng, X.C. Planar Pentacoordinate Carbon in CAl5+: A Global Minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef]
- Guo, J.C.; Feng, L.Y.; Barroso, J.; Merino, G.; Zhai, H.J. Planar or Tetrahedral? A Ternary 17-electron CBe5H4+ Cluster with Planar Pentacoordinate Carbon. Chem. Commun. 2020, 56, 8305–8308. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Parra, L.; Osvaldo Yañez, L.D.; Inostroza, D.; Barroso, J.; Vasquez-Espinal, A.; Merino, G.; Tiznado, W. Planar Hexacoordinate Carbons: Half Covalent, Half Ionic. Angew. Chem. Int. Ed. 2021, 16, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.I.; Li, X.; Wang, L.S. Experimental Observation of Pentaatomic Tetracoordinate Planar Si- and Ge-Containing Molecules: MAl4− and MAl4. Angew. Chem. Int. Ed. 2000, 39, 3307–3310. [Google Scholar] [CrossRef]
- Li, S.D.; Miao, C.Q.; Guo, J.C.; Ren, G.M. Planar Tetra-, Penta-, Hexa-, Hepta-, and Octacoordinate Silicons: A Universal Structural Pattern. J. Am. Chem. Soc. 2004, 126, 16227–16231. [Google Scholar] [CrossRef] [PubMed]
- Minyaev, R.M.; Gribanova, T.N.; Starikov, A.G.; Minkin, V.I. Octacoordinated Main-Group Element Centres in a Planar Cyclic B8 Environment: An Ab Initio Study. Mendeleev Commun. 2001, 11, 213–214. [Google Scholar] [CrossRef]
- Liu, F.L.; Jalbout, A.F. Structural, Electronic, and Magnetic Properties of Heterofullerene C58Si with Odd Number of Atoms and a Near Planar Tetracoordinate Si Atom. J. Mol. Graph. Model. 2008, 26, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Miao, C.Q. M5H5X (M = Ag, Au, Pd, Pt; X = Si, Ge, P, S): Hydrometal Pentagons with D5h Planar Pentacoordinate Nonmetal Centers. J. Phys. Chem. A 2005, 109, 7594–7597. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Ren, G.M.; Miao, C.Q. Hexacoordinate Planar Main Group Atoms Centered in Hexagonal Hydrocopper Complexes Cu6H6X (X = Si, P, As). Inorg. Chem. 2004, 43, 6331–6333. [Google Scholar] [CrossRef]
- Belanzoni, P.; Giorgi, G.; Cerofolini, G.F.; Sgamellotti, A. Planar Tetracoordinated Silicon in Silicon Carbonyl Complexes: A DFT Approach. J. Phys. Chem. A 2006, 110, 4582–4591. [Google Scholar] [CrossRef]
- Guo, J.C.; Li, S.D. Planar Tetra-Coordinate Si and Ge in Perfectly Squared Ni4Cl4X Complexes. J. Mol. Struct. THEOCHEM 2007, 816, 59–65. [Google Scholar] [CrossRef]
- Guo, J.C.; Miao, C.Q.; Ren, G.M. Planar Tetracoordinate Si and Ge in π-aromatic X3Cu3+ (X = Si, Ge) Cations. Comput. Theor. Chem. 2014, 1032, 7–11. [Google Scholar] [CrossRef]
- Guo, J.C.; Wu, H.X.; Ren, G.M.; Miao, C.Q.; Li, Y.X. D3h X3Li3+ (X = C, Si and Ge): Superalkali Cations Containing Three Planar Tetracoordinate X atoms. Comput. Theor. Chem. 2016, 1083, 1–6. [Google Scholar] [CrossRef]
- Wang, M.H.; Dong, X.; Cui, Z.H.; Orozco-Ic, M.; Ding, Y.H.; Barroso, J.; Merino, G. Planar Pentacoordinate Silicon and Germanium Atoms. Chem. Commun. 2020, 56, 13772–13775. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, M.H.; Feng, L.Y.; Zhao, L.Q.; Guo, J.C.; Zhai, H.J.; Cui, Z.H.; Pan, S.; Merino, G. Bare and Ligand Protected Planar Hexacoordinate Silicon in SiSb3M3+ (M = Ca, Sr, Ba) Clusters. Chem. Sci. 2022, 13, 8045–8051. [Google Scholar] [CrossRef] [PubMed]
- Ebner, F.; Greb, L. Calix [4] pyrrole Hydridosilicate: The Elusive Planar Tetracoordinate Silicon Imparts Striking Stability to Its Anionic Silicon Hydride. J. Am. Chem. Soc. 2018, 140, 17409–17412. [Google Scholar] [CrossRef] [PubMed]
- Nukazawa, T.; Iwamoto, T. An Isolable Tetrasilicon Analogue of a Planar Bicyclo [1.1.0]butane with π-Type Single-Bonding Character. J. Am. Chem. Soc. 2020, 142, 9920–9924. [Google Scholar] [CrossRef] [PubMed]
- Ghana, P.; Rump, J.; Schnakenburg, G.; Arz, M.I.; Filippou, A.C. Planar Tetracoordinated Silicon (ptSi): Room-Temperature Stable Compounds Containing Anti-van’t Hoff/Le Bel Silicon. J. Am. Chem. Soc. 2021, 143, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Dong, S.; Yao, S.; Zhu, J.; Driess, M. Synthesis and Reactivity of An Anti-van’t Hoff/Le Bel Compound with a Planar Tetracoordinate Silicon(II) Atom. J. Am. Chem. Soc. 2023, 145, 7084–7089. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Nayhouse, M.J.; Huynh, M.T.; Kuo, J.L.; Melkonian, A.V.; Chavez, G.; Hernando, N.M.; Kowal, M.D.; Liu, C.P. Selected AB42−/− (A = C, Si, Ge; B = Al, Ga, In) Ions: A Battle between Covalency and Aromaticity, and Prediction of Square Planar Si in SiIn42−/−. Phys. Chem. Chem. Phys. 2012, 14, 14815–14821. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ding, Y.H. Pentaatomic Planar Tetracoordinate Silicon with 14 Valence Electrons: A Large-Scale Global Search of SiXnYmq(n + m = 4; q = 0, ±1, −2; X, Y = Main Group Elements From H to Br). J. Comput. Chem. 2015, 36, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Q.; Guo, J.C.; Zhai, H.J. Ternary 14-Electron XB2Be2 (X = Si, Ge, Sn, Pb) Clusters: A Planar Tetracoordinate Silicon (ptSi) System and Its ptGe/Sn/Pb Congeners. Phys. Chem. Chem. Phys. 2022, 24, 7068–7076. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.J.; Xu, J.; Ding, Y.H. A Template for a Planar Tetracoordinate Heavier Group 14 Atom: A Global Study of C2Si2Xq.(X = C, Si, Ge, Sn, Pb; q = +1, 0, −1). Dalton Trans. 2016, 45, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 Energy Evaluation by Direct Methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic Search for Isomers on a Quantum Mechanical Surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.P.; Averkiev, B.B.; Zhai, H.J.; Boldyrev, A.I.; Wang, L.S. All-Boron Analogues of Aromatic Hydrocarbons: B17− and B18−. J. Chem. Phys. 2011, 134, 224304. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic Configuration Interaction. A General Technique for Determining Electron Correlation Energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; Hommes, N.J.R.v.E. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab Initio Classical Trajectories on the Born–Oppenheimer Surface: Hessian-Based Integrators Using Fifth-Order Polynomial and Rational Function Fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Kloda, S.; Kleinpeter, E. Ab Initio Calculation of the Anisotropy Effect of Multiple Bonds and the Ring Current Effect of Arenes—Application in Conformational and Configurational Analysis. J. Chem. Soc. Perkin Trans. 2001, 2, 1893–1898. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.-X.; Guo, J.-C. XB2Bi2 (X = Si, Ge, Sn, Pb): Penta-Atomic Planar Tetracoordinate Si/Ge/Sn/Pb Clusters with 20 Valence Electrons. Int. J. Mol. Sci. 2024, 25, 2819. https://doi.org/10.3390/ijms25052819
Jin Y-X, Guo J-C. XB2Bi2 (X = Si, Ge, Sn, Pb): Penta-Atomic Planar Tetracoordinate Si/Ge/Sn/Pb Clusters with 20 Valence Electrons. International Journal of Molecular Sciences. 2024; 25(5):2819. https://doi.org/10.3390/ijms25052819
Chicago/Turabian StyleJin, Yan-Xia, and Jin-Chang Guo. 2024. "XB2Bi2 (X = Si, Ge, Sn, Pb): Penta-Atomic Planar Tetracoordinate Si/Ge/Sn/Pb Clusters with 20 Valence Electrons" International Journal of Molecular Sciences 25, no. 5: 2819. https://doi.org/10.3390/ijms25052819