
Genetic Characterization of Dilated Cardiomyopathy in Romanian Adult Patients
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics and Gene–Phenotype Correlations
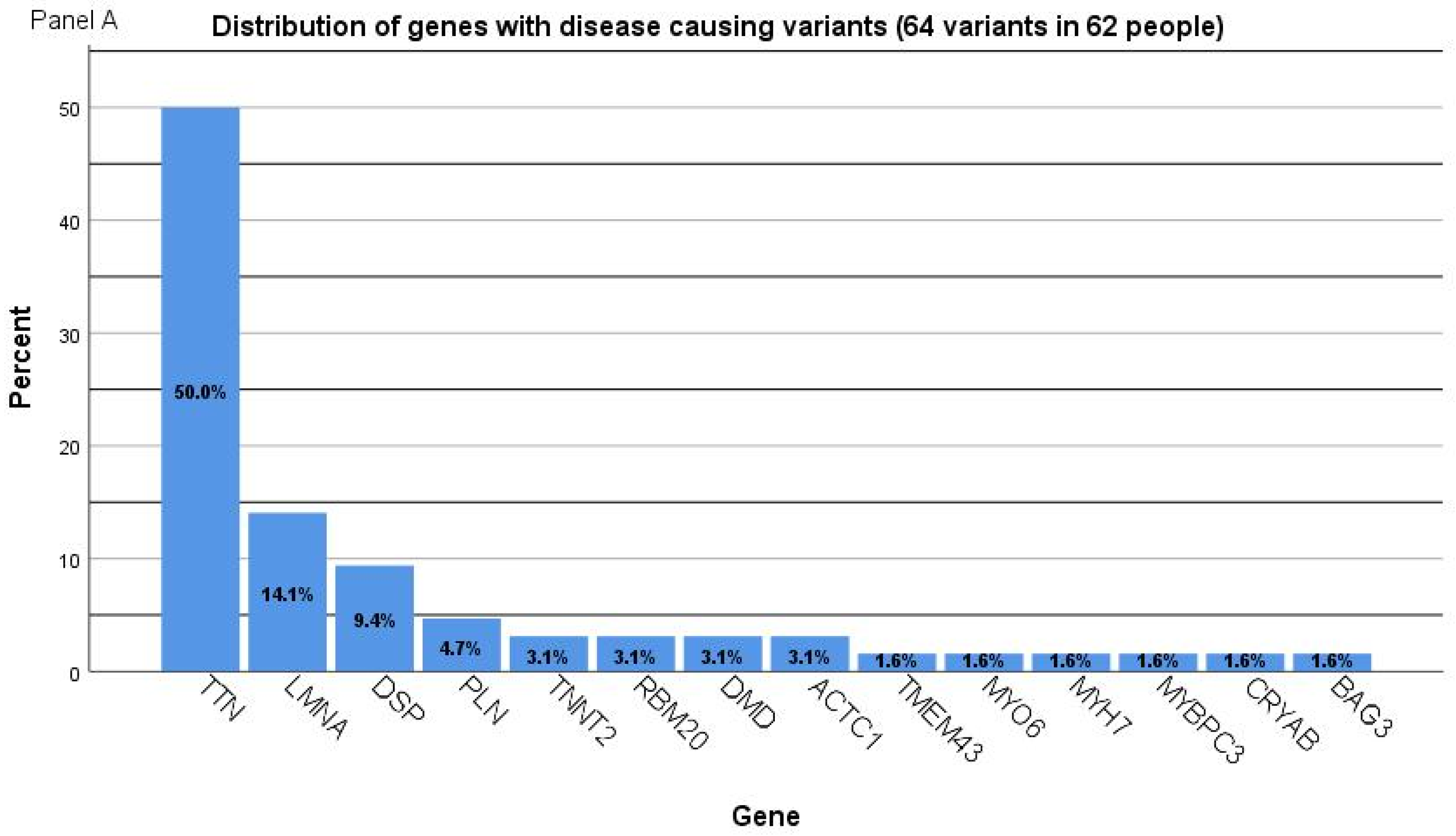
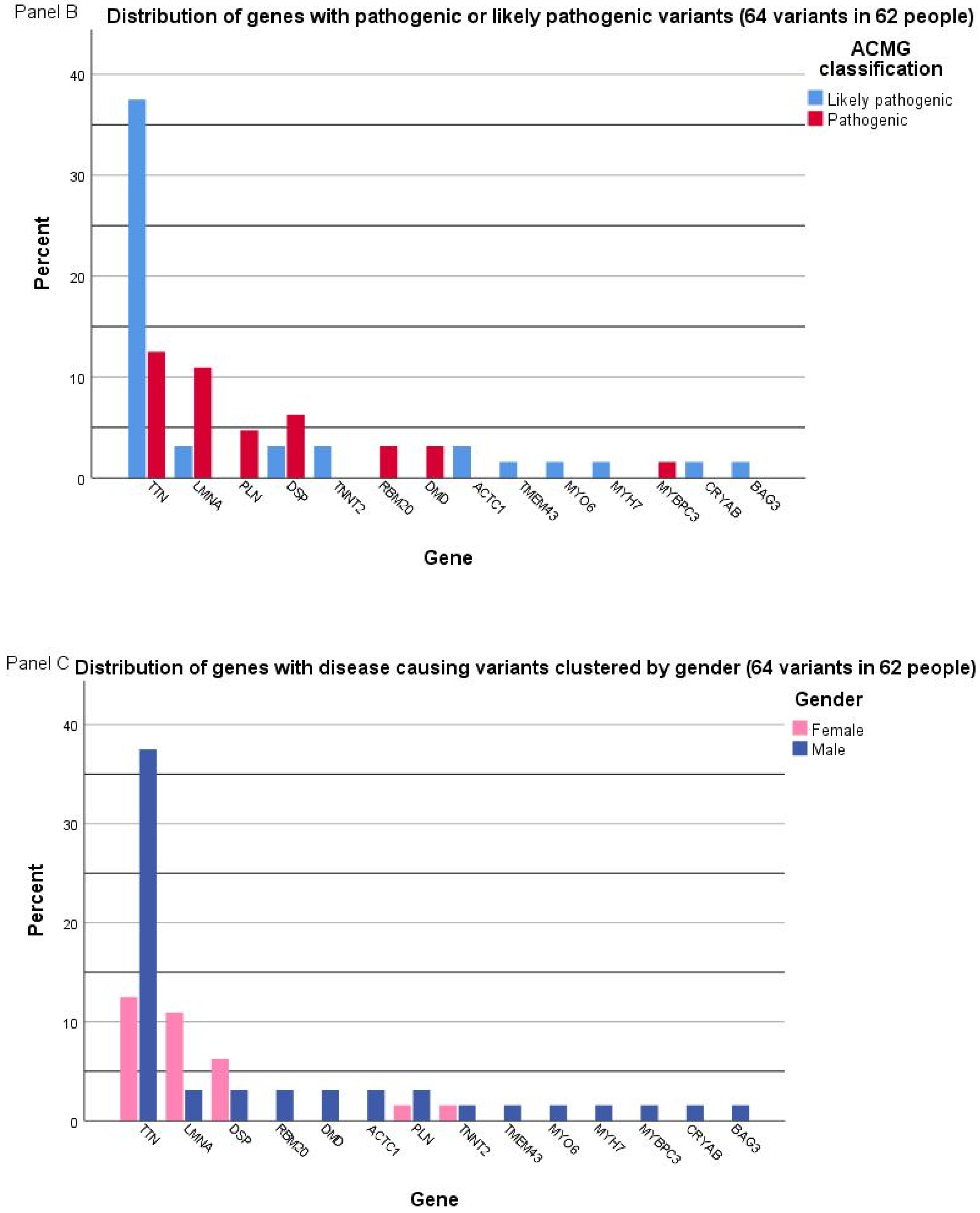
2.2. Genetic Characterization
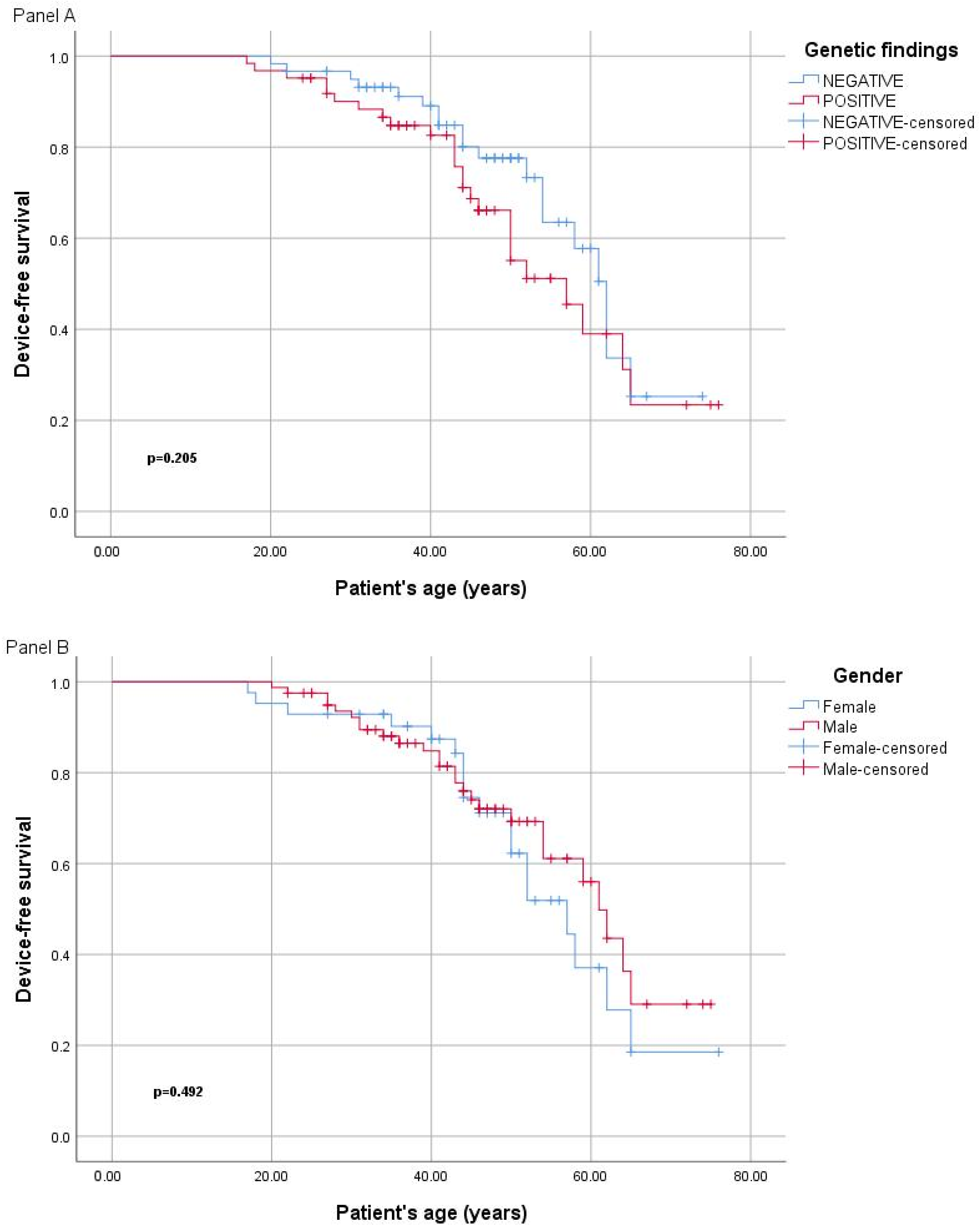
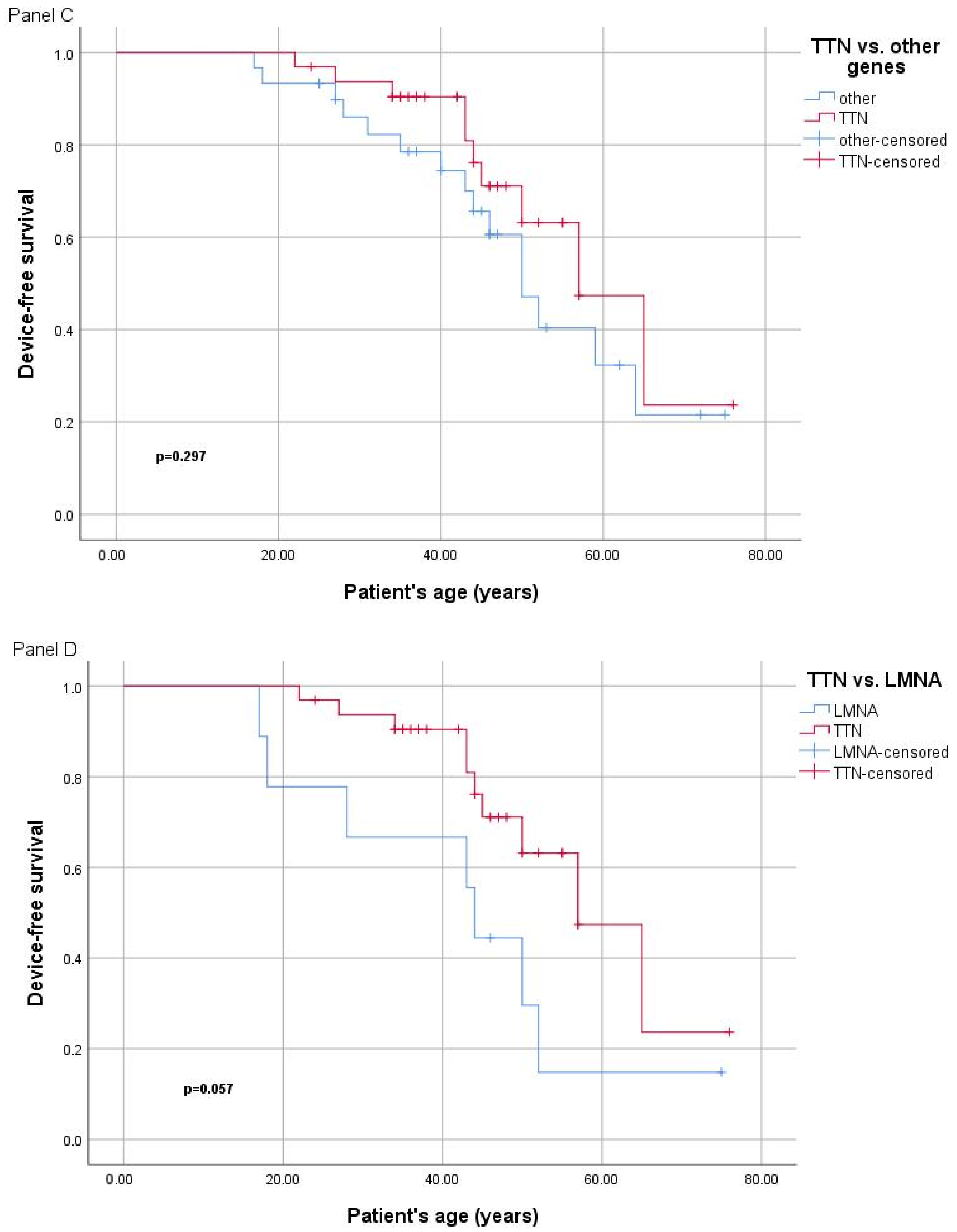
2.3. Genotype–Phenotype Correlation
2.4. Particular Cases in Regards to Molecular Genetics Findings
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Patient Evaluation
4.2. Molecular Diagnostic Tests for DCM
4.3. Variant Interpretation Criteria
4.4. Statistical Analysis
4.5. Ethics Statement
4.6. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
5. Conclusions
- Lay summary:
- Clinical implications:
- Genetic investigations should be considered in sporadic DCM, considering that disease-causing variants were identified in a substantial percentage in this group.
- Family screening, including clinical evaluation and subsequent genetic testing, should be offered to all relatives of index patients with confirmed familial DCM.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | The American College of Medical Genetics and Genomics |
| AMP | Association for Molecular Pathology |
| B | Benign |
| BNP | brain natriuretic peptides |
| BSA | body surface area |
| BWA | Burrows–Wheeler alignment |
| CMR | cardiac magnetic resonance |
| CRTD | Cardiac Resynchronization Therapy Devices |
| CRTP | cardiac resynchronization therapy with pacemaker |
| DCM | dilated cardiomyopathy |
| ECG | Electrocardiogram |
| HF | heart failure |
| ICD | implantable cardioverter-defibrillator |
| LB | likely benign |
| LP | likely pathogenic |
| LVED | left ventricular end-diastolic |
| LVEF | left ventricular ejection fraction |
| NGS | next-generation sequencing |
| P | Pathogenic |
| SCD | sudden cardiac death |
| SD | standard deviation |
| VUS | variant of uncertain significance |
| WGS | whole-genome sequencing |
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Kinnamon, D.D.; Jordan, E.; Platt, J.; Vatta, M.; Dorschner, M.O.; Starkey, C.A.; Mead, J.O.; Ai, T.; Burke, W.; et al. Variant Interpretation for Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, e002480. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Cardiac and Vascular Biology; Springer: Cham, Switzerland, 2019; Volume 7. [Google Scholar]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. J. Arrhythm. 2022, 24, 1307–1367. [Google Scholar]
- Jurcut, R.; Fetecău, B. Genetic testing for cardiomyopathies-when science and health policies join in personalizing cardiovascular prevention. Eur. J. Prev. Cardiol. 2022, 29, 1785–1788. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef]
- Asselbergs, F.W.; Sammani, A.; Elliott, P.; Gimeno, J.R.; Tavazzi, L.; Tendera, M.; Kaski, J.P.; Maggioni, A.P.; Rubis, P.P.; Jurcut, R.; et al. Differences between familial and sporadic dilated cardiomyopathy: ESC EORP Cardiomyopathy & Myocarditis registry. ESC Heart Fail. 2020, 8, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Vu, M.T.T.; Do, T.N.P.; Tran, T.H.N.; Do, T.H.; Nguyen, T.M.H.; Huynh, B.N.T.; Le, L.A.; Pham, N.T.N.; Nguyen, T.D.A.; et al. Genetic Determinants and Genotype-Phenotype Correlations in Vietnamese Patients with Dilated Cardiomyopathy. Circ. J. 2021, 85, 1469–1478. [Google Scholar] [CrossRef]
- Akinrinade, O.; Ollila, L.; Vattulainen, S.; Tallila, J.; Gentile, M.; Salmenperä, P.; Koillinen, H.; Kaartinen, M.; Nieminen, M.S.; Myllykangas, S.; et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur. Heart J. 2015, 36, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Cannata, A.; Merlo, M.; Manca, P.; Ferro, M.D.; Paldino, A.; Artico, J.; Gentile, P.; Jirikowic, J.; Todd, E.; Salcedo, E.; et al. The late-onset dilated cardiomyopathy. Eur. Heart J. 2020, 41, ehaa946.2096. [Google Scholar] [CrossRef]
- Verdonschot, J.A.; Hazebroek, M.R.; Krapels, I.P.; Henkens, M.T.; Raafs, A.; Wang, P.; Merken, J.J.; Claes, G.R.; Vanhoutte, E.K.; Wijngaard, A.v.D.; et al. Implications of Genetic Testing in Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 476–487. [Google Scholar] [CrossRef]
- Zhang, X.-L.; Xie, J.; Lan, R.-F.; Kang, L.-N.; Wang, L.; Xu, W.; Xu, B. Genetic Basis and Genotype–Phenotype Correlations in Han Chinese Patients with Idiopathic Dilated Cardiomyopathy. Sci. Rep. 2020, 10, 2226. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Hoorntje, E.T.; de Brouwer, R.; Oomen, A.; Amin, A.; van der Heijden, J.F.; Karper, J.C.; Westenbrink, B.D.; Silljé, H.H.W.; te Riele, A.S.J.M.; et al. Rationale and design of the PHOspholamban RElated CArdiomyopathy intervention STudy (i-PHORECAST). Neth. Heart J. 2022, 30, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Crasto, S.; My, I.; Di Pasquale, E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 2020, 11, 761. [Google Scholar] [CrossRef]
- Yeung, C.; Enriquez, A.; Suarez-Fuster, L.; Baranchuk, A. Atrial fibrillation in patients with inherited cardiomyopathies. EP Eurospace 2019, 21, 22–32. [Google Scholar] [CrossRef]
- Goodyer, W.R.; Dunn, K.; Caleshu, C.; Jackson, M.; Wylie, J.; Moscarello, T.; Platt, J.; Reuter, C.; Smith, A.; Trela, A.; et al. Broad Genetic Testing in a Clinical Setting Uncovers a High Prevalence of Titin Loss-of-Function Variants in Very Early Onset Atrial Fibrillation. Circ. Genom. Precis. Med. 2019, 12, e002713. [Google Scholar] [CrossRef]
- Andreasen, L.; Bertelsen, L.; Ghouse, J.; Lundegaard, P.R.; Ahlberg, G.; Refsgaard, L.; Rasmussen, T.B.; Eiskjær, H.; Haunsø, S.; Vejlstrup, N.; et al. Early-onset atrial fibrillation patients show reduced left ventricular ejection fraction and increased atrial fibrosis. Sci. Rep. 2020, 10, 10039. [Google Scholar] [CrossRef]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L.; Roden, D.M. Cardiac Sodium Channel (SCN5A) Variants Associated with Atrial Fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef]
- Tayal, U.; Prasad, S.; Cook, S.A. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017, 9, 20. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Molina, M.S.; Restrepo-Cordoba, M.A.; Ferro, M.D.; Stolfo, D.; Johnson, R.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T.J.; Ferguson, D.A.; Brodsky, G.L.; et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.W.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail. 2013, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef]
- Nishiuchi, S.; Makiyama, T.; Aiba, T.; Nakajima, K.; Hirose, S.; Kohjitani, H.; Yamamoto, Y.; Harita, T.; Hayano, M.; Wuriyanghai, Y.; et al. Gene-Based Risk Stratification for Cardiac Disorders in LMNA Mutation Carriers. Circ. Cardiovasc. Genet. 2017, 10, e001603. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef]
- Hof, I.E.; van der Heijden, J.F.; Kranias, E.G.; Sanoudou, D.; de Boer, R.A.; van Tintelen, J.P.; van der Zwaag, P.A.; Doevendans, P.A. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth. Heart J. 2019, 27, 64–69. [Google Scholar] [CrossRef]
- Roncarati, R.; Anselmi, C.V.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef]
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P.; BTech, K.W.; Statham, A.L.; Lundie, B.; Bagnall, R.D.; et al. Genome Sequencing as a First-Line Genetic Test in Familial Dilated Cardiomyopathy. Genet. Med. 2019, 21, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Lesurf, R.; Said, A.; Akinrinade, O.; Breckpot, J.; Delfosse, K.; Liu, T.; Yao, R.; Persad, G.; McKenna, F.; Noche, R.R.; et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. NPJ Genom. Med. 2022, 7, 18. [Google Scholar] [CrossRef]
- Wilsbacher, L.D. Clinical Implications of the Genetic Architecture of Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2020, 22, 170. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 233–271. [Google Scholar] [CrossRef]
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef]
- Lincoln, S.E.; Truty, R.; Lin, C.-F.; Zook, J.M.; Paul, J.; Ramey, V.H.; Salit, M.; Rehm, H.L.; Nussbaum, R.L.; Lebo, M.S. A Rigorous Interlaboratory Examination of the Need to Confirm Next-Generation Sequencing–Detected Variants with an Orthogonal Method in Clinical Genetic Testing. J. Mol. Diagn. 2019, 21, 318–329. [Google Scholar] [CrossRef]
- Chirita-Emandi, A.; Andreescu, N.; Zimbru, C.G.; Tutac, P.; Arghirescu, S.; Serban, M.; Puiu, M. Challenges in reporting pathogenic/potentially pathogenic variants in 94 cancer predisposing genes—In pediatric patients screened with NGS panels. Sci. Rep. 2020, 10, 223. [Google Scholar] [CrossRef]
- Chirita-Emandi, A.; Andreescu, N.; Popa, C.; Mihailescu, A.; Riza, A.-L.; Plesea, R.; Ioana, M.; Arghirescu, S.; Puiu, M. Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J. Med. Genet. 2020, 58, 648–652. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All n = 122 | Disease-Causing Findings n = 62 | Negative & VUS n = 60 | p Value | Familial n = 64 | Sporadic n = 58 | p Value |
|---|---|---|---|---|---|---|---|
| Gender male n (%) | 79 (66.6%) | 41 (66.6%) | 39 (65.5%) | 0.524 | 35 (54.6%) | 45 (77.6%) | 0.006 |
| Age, years (mean ± SD) | 45.8 ± 12.8 | 44.9 ± 13.1 | 46.8 ± 12.4 | 0.415 | 43.5 ± 12.0 | 48.3 ± 13.2 | 0.039 |
| Age at diagnosis (mean ± SD) | 41.4 ± 12.3 | 40.2 ± 12.5 | 42.7 ± 12.1 | 0.277 | 38.7 ± 11.4 | 44.4 ± 12.8 | 0.011 |
| Presentation reason n (%) | |||||||
| Cardiac symptoms | 101 (82.3%) | 48 (80.0%) | 53 (88.3%) | 0.191 | 48 (75%) | 53 (91.4%) | 0.011 |
| Sudden cardiac death episode | 3 (2.4%) | 2 (3.2%) | 1 (1.7%) | 0.512 | 1 (1.5%) | 2 (3.4%) | 0.463 |
| Incidental discovery | 8(6.5%) | 3 (4.8%) | 5(8.3%) | 0.389 | 4 (6.2%) | 4 (6.9%) | 0.011 |
| Family screening | 10 (8.1%) | 8 (12.9%) | 2(3.3%) | 0.122 | 10 (15.6%) | 0 | 0.012 |
| Heart failure features | |||||||
| LVEF (mean ± SD) | 35.3 ± 11.1 | 36.2 ± 11.7 | 34.3 ± 10.4 | 0.343 | 36.6 ± 10.9 | 33.8 ± 11.2 | 0.168 |
| LVEF ≤ 35% n (%) | 59 (48.3%) | 26 (41.9%) | 33 (55%) | 0.103 | 28 (43.7%) | 31 (53.4%) | 0.187 |
| NTproBNP (pg/mL) n = 63 | 3357.1 ± 442.4 | 3967.09 ± 4971.13 | 2642.03 ± 3649.22 | 0.062 | 3053.86 ± 4029.21 | 3653.86 ± 4853.17 | 0.126 |
| BNP (pg/mL) n = 39 | 435.6 ± 744.7 | 662.9 ± 974.0 | 219.7 ± 331.1 | 0.073 | 275.2 ± 554.7 | 722.0 ± 956.6 | 0.072 |
| Complications n (%) | |||||||
| Ventricular arrhythmias | 35 (28.6%) | 20 (32.2%) | 15 (25%) | 0.407 | 17 (26.5%) | 20 (34.5%) | 0.204 |
| Atrial fibrillation | 23 (18.8%) | 16 (25.8%) | 7 (11.6%) | 0.041 | 12 (18.7%) | 11 (18.9%) | 0.599 |
| Heart conduction disorder | 41 (33.3%) | 17 (27.4%) | 24 (40%) | 0.159 | 19 (29.7%) | 22 (37.9%) | 0.175 |
| Fibrosis on CMR | 54/73 (73.9%) | 31/41 (75.6%) | 23/32 (71.8%) | 0.461 | 30/38 (78.9%) | 24/35 (80.6%) | 0.229 |
| Heart transplantation | 1 (0.8%) | 1 (1.6%) | 0 | 0.529 | 1 (1.6%) | 0 | 0.529 |
| Deceased | 5 (4.1%) | 3 (4.8%) | 2 (3.3%) | 0.508 | 4 (6.2%) | 1 (1.7%) | 0.215 |
| Devices | |||||||
| Pacemaker n (%) | 6 (4.9%) | 3 (4.8%) | 3 (5%) | 0.644 | 3 (4.6%) | 3 (5.2%) | 0.612 |
| Any ICD (ICD + CRT-D) n (%) | 36 (29.3%) | 22 (35.4%) | 14 (23.3%) | 0.124 | 21 (32.8%) | 15 (25.8%) | 0.352 |
| CRTP n (%) | 6 (4.9%) | 2 (3.2%) | 4 (6.6%) | 0.324 | 1 (1.6%) | 5 (86.2%) | 0.083 |
| Age at ICD implant n = 35 (mean ± SD) | 40.97 ± 13.58 | 39.29 ± 13.53 | 43.50 ± 13.74 | 0.379 | 39.40 ± 13.41 | 43.07 ± 13.98 | 0.441 |
| Age at CRT implant n = 14 (mean ± SD) | 49.43 ± 13.09 | 50.20 ± 6.79 | 49.00 ± 15.96 | 0.877 | 47.75 ± 19.19 | 50.10 ± 11.09 | 0.830 |
| Familial or Sporadic | Sex | Age at Diagnosis (Years) | Gene | NM Number | Transcript (Protein) | Zygosity | Classify-Cation | Variant Type |
|---|---|---|---|---|---|---|---|---|
| familial | M | 32 | ACTC1 | NM_005159.5 | c.854T>A (p.Met285Lys) | het | LP (VUS) | missense * |
| familial | M | 47 | ACTC1 | NM_005159.5 | c.854T>A (p.Met285Lys) | het | LP (VUS) | missense * |
| familial | M | 39 | BAG3 | NM_004281.4 | c.920dupC (p.H308Pfs) | het | LP | frameshift * |
| sporadic | M | 56 | CRYAB | NM_001289808.2 | c.166C>T (p.Arg56Trp) | het | LP | missense |
| familial | M | 17 | DMD | NM_004006.3 | c.10801C>T (p.Gln3601Ter) | het | P | nonsense * |
| sporadic | M | 20 | DMD | NM_004006.3 | c.6913-?_7200+?del | het | P | frameshift |
| sporadic | F | 41 | DSP | NM_004415.4 | c.(2630+1_2631-1)_(2877+2878-1)del | het | LP | frameshift |
| sporadic | F | 51 | DSP | NM_004415.2 | c.2498dup (p.Lys834Glufs) | het | P | frameshift |
| familial | F | 38 | DSP | NM_004415.2 | c.313C>T (p.Arg105Ter) | het | P | nonsense |
| familial | M | 42 | DSP | NM_004415.2 | c.313C>T (p.Arg105Ter) | het | P | nonsense |
| sporadic | F | 41 | DSP | NM_004415.2 | c.5212C>T (p.Arg1738Ter) | het | P | nonsense |
| sporadic | M | 70 | DSP | NM_004415.2 | c.597+1G>A | het | LP | splicesite |
| familial | F | 50 | LMNA | NM_170707.4 | c.1003C>T (p.Arg335Trp) | het | P | missense |
| familial | M | 28 | LMNA | NM_170707.4 | c.1003C>T p.(Arg335Trp) | het | P | missense |
| familial | F | 19 | LMNA | NM_170707.3 | c.448A>C (p.Thr150Pro) | het | P | missense |
| familial | F | 16 | LMNA | NM_170707.3 | c.448A>C (p.Thr150Pro) | het | LP | missense |
| familial | F | 44 | LMNA | NM_170707.3 | c.604G>T (p.Glu202Ter) | het | P | nonsense |
| sporadic | F | 45 | LMNA | NM_170707.3 | c.673C>T (p.Arg225Ter) | het | P | nonsense |
| sporadic | M | 68 | LMNA | NM_170707.3 | c.886C>T (p.Arg296Cys) | het | LP (VUS) | missense |
| familial | F | 40 | LMNA | NM_170707.3 | c.980_995del (p.Leu327Profs) | het | P | frameshift |
| familial | F | 41 | LMNA | NM_170707.3 | c.980_995del (p.Leu327Profs) | het | P | frameshift |
| $ sporadic | M | 64 | MYBPC3 | NM_000256.3 | c.1504C>T (p.Arg502Trp) | het | P | missense |
| sporadic | M | 24 | MYH7 | NM_000257.4 | c.2458G>A (p.Ala820Thr) | het | LP | missense * |
| sporadic | M | 31 | MYO6 | NM_004999.3 | c.755G>A (p.Cys252Tyr) | het | LP (VUS) | missense * |
| $ sporadic | M | 64 | PLN | NM_002667.3 | c.116T>G (p.Leu39Ter) | het | P | nonsense |
| # sporadic | M | 44 | PLN | NM_002667.3 | c.116T>G (p.Leu39Ter) | het | P | missense |
| familial | F | 30 | PLN | NM_002667.3 | c.116T>G (p.Leu39Ter) | hom | P | missense |
| familial | M | 31 | RBM20 | NM_001134363.2 | c.1913C>T (p.Pro638Leu) | het | P | missense |
| sporadic | M | 34 | RBM20 | NM_001134363.3 | c.2737G>A (p.Glu913Lys) | het | P | missense |
| ^ familial | M | 20 | RYR1 | NM_000540.2 | Deletion (Exons 48-49) | het | P | frameshift |
| sporadic | M | 27 | TMEM43 | NM_024334.2 | c.718C>A (p.Arg240Ser) | het | LP (VUS) | missense * |
| familial | F | 49 | TNNT2 | NM_001001430.2 | c.400C>T (p.Arg134Trp) | het | LP | missense |
| sporadic | M | 42 | TNNT2 | NM_001001430.2 | c.517C>T (p.Arg173Trp) | het | LP | missense |
| familial | F | 30 | TTN | NM_001267550.2 | c.101418del (p.Ala33807HisfsTer) | het | LP | frameshift * |
| sporadic | M | 40 | TTN | NM_001267550.2 | c.101793_101794del (p.His33931GlnfsTer) | het | LP | frameshift |
| familial | M | 31 | TTN | NM_001267550.2 | c.107635C>T (p.Gln35879Ter) | het | LP | nonsense |
| sporadic | M | 65 | TTN | NM_001267550.2 | c.46986dup (p.Asn15663Ter) | het | P | frameshift * |
| ^ familial | M | 20 | TTN | NM_001267550.2 | c.46986dup (p.Asn15663Ter) | het | P | frameshift * |
| familial | F | 65 | TTN | NM_001267550.2 | c.60733C>T (p.Arg20245Ter) | het | P | nonsense |
| familial | M | 52 | TTN | NM_001267550.2 | c.60733C>T (p.Arg20245Ter) | het | P | nonsense |
| familial | F | 45 | TTN | NM_001267550.2 | c.68269del (p.His22757ThrfsTer) | het | LP | frameshift * |
| familial | M | 45 | TTN | NM_001267550.2 | c.68575_68576dup (p.Ile22861SerfsTer) | het | LP | frameshift * |
| familial | M | 50 | TTN | NM_001267550.2 | c.68575_68576dup (p.Ile22861SerfsTer) | het | LP | frameshift * |
| sporadic | M | 41 | TTN | NM_001267550.2 | c.68824del (p.Glu22942ArgfsTer) | het | LP | frameshift |
| sporadic | M | 55 | TTN | NM_001267550.2 | c.70437del (p.Lys23480AsnfsTer) | het | LP | frameshift * |
| familial | M | 39 | TTN | NM_001267550.2 | c.70437del (p.Lys23480AsnfsTer) | het | LP | frameshift * |
| sporadic | M | 32 | TTN | NM_001267550.2 | c.70978C>T (p.Arg23660Ter) | het | P | nonsense |
| familial | M | 33 | TTN | NM_001267550.2 | c.73646C>G (p.Ser24549Ter) | het | LP | nonsense * |
| sporadic | F | 54 | TTN | NM_001267550.2 | c.73646C>G (p.Ser24549Ter) | het | LP | nonsense * |
| sporadic | M | 36 | TTN | NM_001267550.2 | c.74338C>T (p.Arg24780Ter) | het | P | nonsense |
| familial | M | 39 | TTN | NM_001267550.2 | c.74338C>T (p.Arg24780Ter) | het | P | nonsense |
| familial | F | 55 | TTN | NM_001267550.2 | c.79273A>T (p.Lys26425Ter) | het | LP | nonsense * |
| familial | F | 34 | TTN | NM_001267550.2 | c.79273A>T (p.Lys26425Ter) | het | LP | nonsense * |
| familial | M | 28 | TTN | NM_001267550.2 | c.82172G>A (p.Trp27391Ter) | het | LP | nonsense * |
| familial | F | 48 | TTN | NM_001267550.2 | c.82172G>A (p.Trp27391Ter) | het | LP | nonsense * |
| familial | M | 47 | TTN | NM_001267550.2 | c.82415_82419dup (p.Ser27474LeufsTer) | het | LP | frameshift * |
| familial | M | 46 | TTN | NM_001267550.2 | c.82415_82419dup (p.Ser27474LeufsTer) | het | LP | frameshift * |
| familial | F | 35 | TTN | NM_001267550.2 | c.89882_89885del (p.Gly29961AspfsTer) | het | LP | frameshift * |
| sporadic | M | 32 | TTN | NM_001267550.2 | c.92294del (p.Arg30765AsnfsTer3) | het | LP | frameshift * |
| sporadic | M | 21 | TTN | NM_001267550.2 | c.92317C>T (p.Arg30773Ter) | het | P | nonsense |
| sporadic | M | 33 | TTN | NM_001267550.2 | c.94996_95008del (p.Tyr31666GlufsTer) | het | LP | frameshift * |
| sporadic | M | 36 | TTN | NM_001267550.2 | c.95055del (p.Lys31685AsnfsTer4) | het | LP | frameshift * |
| familial | M | 43 | TTN | NM_001267550.2 | c.59626+1G>A | het | LP | splicesite * |
| familial | M | 43 | TTN | NM_001267550.2 | c.71184del (Pro23729HisfsTer16) | het | LP | frameshift * |
| # sporadic | M | 44 | TTN | NM_001267550.2 | c.94128del (p.Lys31375_Tyr31376insTer) | het | LP | frameshift * |
| Variable | All Positive n = 62 | TTN n = 32 | Other Genes n = 30 | p Value TTN/Other | LMNA n = 9 | p Value TTN/LMNA |
|---|---|---|---|---|---|---|
| Gender male n (%) | 41 (66.1) | 24 (75%) | 17 (56.7%) | 0.104 | 2 (22.2%) | 0.006 |
| Age, years (mean ± SD) | 44.9 ± 13.1 | 45.1 ± 11.6 | 44.7 ± 14.8 | 0.915 | 44.1 ± 17.3 | 0.847 |
| Age at diagnosis (mean ± SD) | 40.2 ± 12.6 | 41.2 ± 10.9 | 39.2 ± 14.1 | 0.550 | 39.0 ± 16.1 | 0.880 |
| Presentation reason n (%) | ||||||
| Cardiac symptoms | 49 (79%) | 24 (75%) | 25 (83.3%) | 0.679 | 8 (88.9%) | 0.350 |
| Sudden cardiac death episode | 2 (3.2%) | 1 (3.1%) | 1 (3.3%) | 0.738 | 0 | 0.780 |
| Incidental discovery | 4 (6.4%) | 2 (6.2%) | 2 (6.6%) | 0.910 | 0 | 0.427 |
| Family screening no symptoms | 8 (12.9%) | 5 (15.6%) | 3 (10.0%) | 0.645 | 1 (1.1%) | 0.176 |
| Heart failure | ||||||
| LVEF value (mean ± SD) | 36.2 ± 11.7 | 34.9 ± 11.7 | 37.6 ± 11.7 | 0.359 | 41.9 ± 9.2 | 0.073 |
| LVEF ≤ 35% n (%) | 26 (41.9%) | 15 (46.9%) | 11 (36.3%) | 0.289 | 2 (22.2%) | 0.174 |
| NTproBNP (pg/mL) (mean ± SD) | 3967.1 ± 4971.0 (n = 34) | 3695.3 ± 5083.2 (n = 21) | 4406.0 ± 4955.4 (n = 13) | 0.691 | 5533.6 ± 6256.4 (n = 6)) | 0.464 |
| BNP (pg/mL) (mean ± SD) | 662.8 ± 974.0 (n = 19) | 33.5 ± 41.7 (n = 4) | 830.6 ± 1037.3 (n = 15) | 0.010 | 659.0 ± 729.5 (n = 4) | 0.185 |
| Complications n (%) | ||||||
| Ventricular arrhythmias | 17 (27.4%) | 9 (28.1%) | 3 (10%) | 0.293 | 4 (44.4%) | 0.294 |
| Atrial fibrillation | 16 (25.8%) | 7 (21.9%) | 9 (30%) | 0.301 | 5 (55.6%) | 0.064 |
| Heart conduction disorder | 17 (27.4%) | 6 (18.8%) | 11 (36.7) | 0.097 | 6 (66.7%) | 0.011 |
| Fibrosis on MRI | 31/41 (76.5%) | 16/21 (76.2%) | 16/20 (80.0%) | 0.463 | 8/8 (100%) | 0.126 |
| Heart transplantation | 1 (1.6%) | 0 | 1 (3.3%) | 0.475 | 1 (11.1%) | 0.220 |
| Deceased | 3 (4.8%) | 2 (6.3%) | 1 (3.3%) | 0.525 | 0 | 0.605 |
| Devices | ||||||
| Pacemaker n (%) | 3 (4.8%) | 1 (3.1%) | 2 (6.7%) | 0.475 | 1 (11.1%) | 0.395 |
| Any ICD (ICD + CRT-D) n (%) | 22 (35.5%) | 9 (28.1%) | 13 (43.3%) | 0.115 | 6 (66.7%) | 0.022 |
| CRTP n (%) | 3 (4.8%) | 1 | 2 (6.7) | 0.230 | 1 (11.1%) | 0.220 |
| Age at ICD implant (mean ± SD) | 39.3 ± 13.5 | 42.22 ± 13.49 | 37.1 ± 13.7 | 0.403 | 33.3 ± 14.2 | 0.243 |
| Age at CRT implant (mean ± SD) | 50.0 ± 6.8 | 50 | 50.2 ± 7.8 | 0.979 | 52 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voinescu, O.R.; Ionescu, B.I.; Militaru, S.; Afana, A.S.; Sascau, R.; Vasiliu, L.; Onciul, S.; Dobrescu, M.A.; Cozlac, R.A.; Cozma, D.; et al. Genetic Characterization of Dilated Cardiomyopathy in Romanian Adult Patients. Int. J. Mol. Sci. 2024, 25, 2562. https://doi.org/10.3390/ijms25052562
Voinescu OR, Ionescu BI, Militaru S, Afana AS, Sascau R, Vasiliu L, Onciul S, Dobrescu MA, Cozlac RA, Cozma D, et al. Genetic Characterization of Dilated Cardiomyopathy in Romanian Adult Patients. International Journal of Molecular Sciences. 2024; 25(5):2562. https://doi.org/10.3390/ijms25052562
Chicago/Turabian StyleVoinescu, Oana Raluca, Bogdana Ioana Ionescu, Sebastian Militaru, Andreea Sorina Afana, Radu Sascau, Laura Vasiliu, Sebastian Onciul, Mihaela Amelia Dobrescu, Ramona Alina Cozlac, Dragos Cozma, and et al. 2024. "Genetic Characterization of Dilated Cardiomyopathy in Romanian Adult Patients" International Journal of Molecular Sciences 25, no. 5: 2562. https://doi.org/10.3390/ijms25052562