
Current Understanding of Immune Thrombocytopenia: A Review of Pathogenesis and Treatment Options

Abstract
:1. Introduction
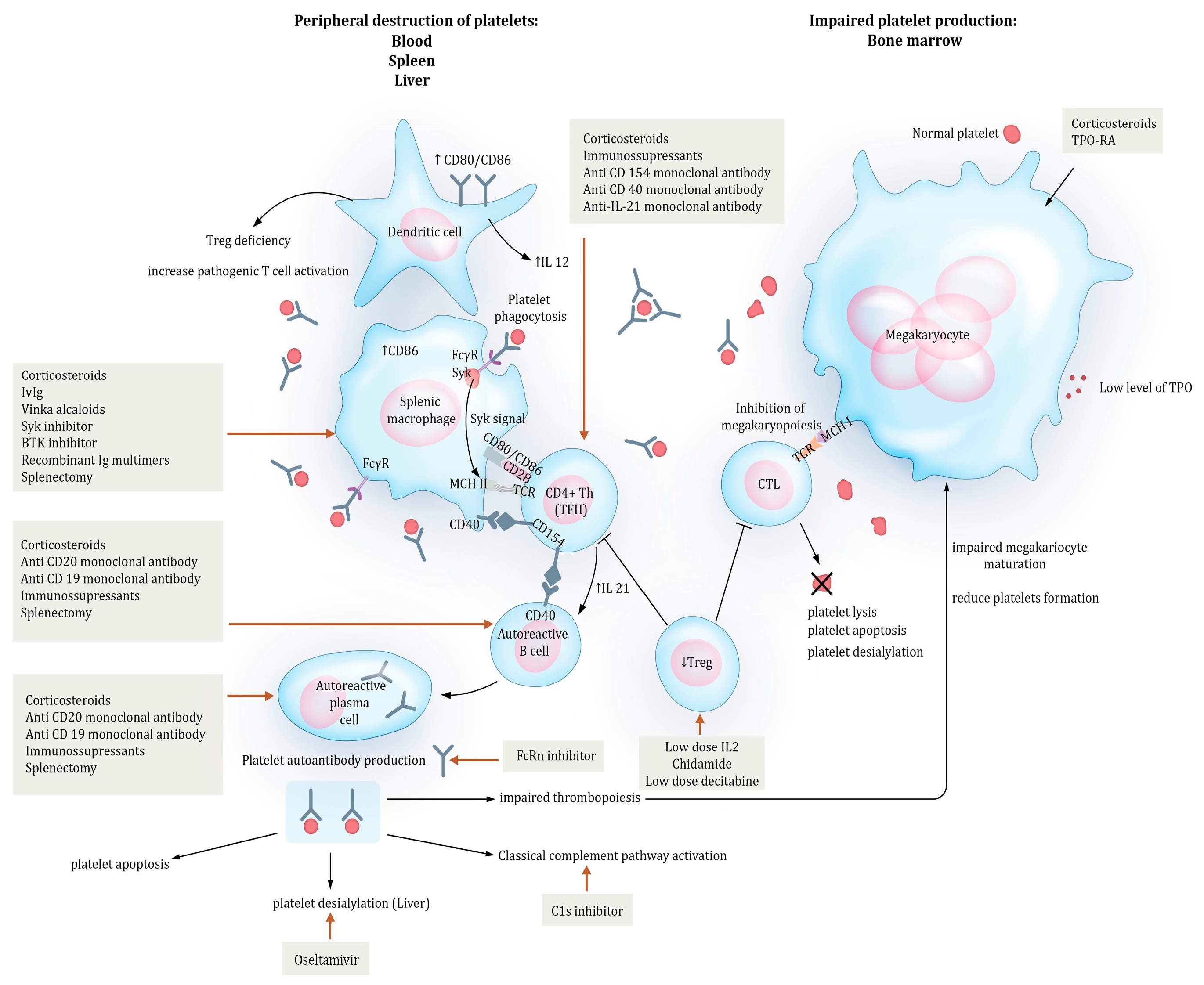
2. Pathogenesis
2.1. Mechanisms of Peripheral Destruction
2.2. T-Cell Involvement
2.3. Impaired Thrombopoiesis
3. The Role of Platelets in ITP
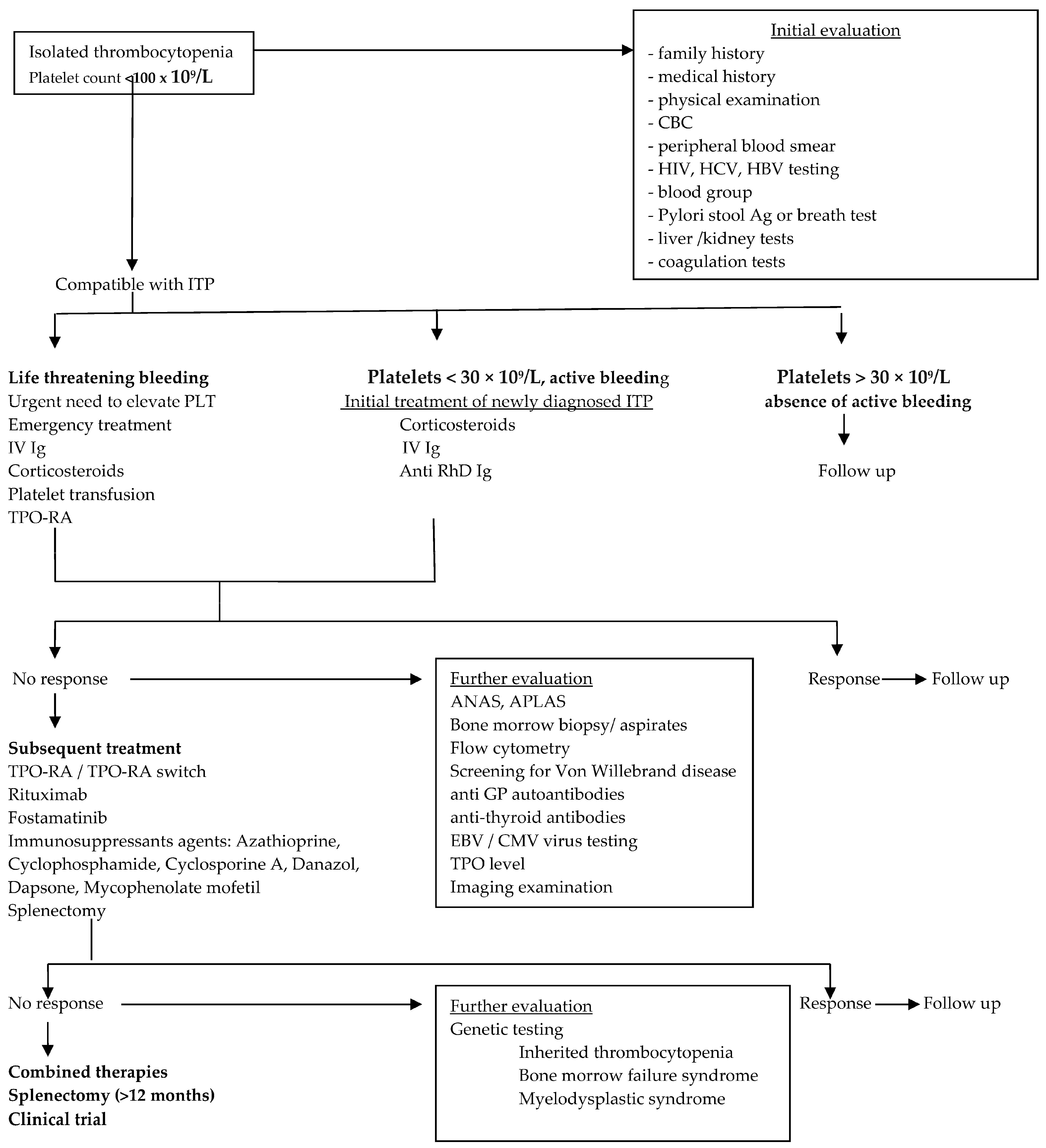
4. Current Management of ITP
4.1. Whom to Treat
4.2. Bleeding Risk
4.3. How to Treat
5. New Therapeutic Targets in ITP: From Pathophysiology to Treatment
5.1. Inhibition of Phagocytosis by Targeting FCγR Transduction Signal
5.1.1. Inhibition of Complement
5.1.2. Inhibition of Platelet Desialylation
5.2. B-Cell- and Plasma Cell-Targeting Therapies
5.2.1. Anti-CD20-Targeting Therapies
5.2.2. Combination Therapies
5.2.3. Plasma Cell-Targeting Therapies
5.3. T Cell-Targeting Therapies
5.3.1. CD40/CD154 Blockage
5.3.2. IL-21 Inhibition
5.3.3. Treg Restoration
5.4. Enhancement of Thrombopoiesis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rodeghiero, F.; Stasi, R.; Gernsheimer, T.; Michel, M.; Provan, D.; Arnold, D.M.; Bussel, J.B.; Cines, D.B.; Chong, B.H.; Cooper, N.; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009, 113, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Comont, T.; Adoue, D. New insights into the epidemiology of immune thrombocytopenia in adult patients: Impact for clinical practice. La Rev. De Med. Interne 2021, 42, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, P.E.; Hall, S.A.; Feudjo-Tepie, M.; Mitrani-Gold, F.S.; Logie, J. The incidence of idiopathic thrombocytopenic purpura among adults: A population-based study and literature review. Eur. J. Haematol. 2009, 83, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Terrell, D.R.; Beebe, L.A.; Vesely, S.K.; Neas, B.R.; Segal, J.B.; George, J.N. Critical Review The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol. 2010, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.; Cooper, N.; Boccia, R.; Zaja, F.; Newland, A. Immune thrombocytopenia. Expert Rev. Hematol. 2021, 14, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Blanchette, V.S. Immune thrombocytopenic purpura. N. Engl. J. Med. 2002, 346, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Liebman, H.; Stasi, R. Pathobiology of secondary immune thrombocytopenia. Semin. Hematol. 2009, 46, S2–S14. [Google Scholar] [CrossRef]
- Ayesh, M.H.; Yousef, H.; Alawneh, K.; Khassawneh, B.; Khader, Y.; Kasasbeh, A. Adult Primary and Secondary Immune Thrombocytopenic Purpura: A Comparative Analysis of Characteristics and Clinical Course. Clin. Appl. Thromb./Hemost. 2013, 19, 327–330. [Google Scholar] [CrossRef]
- Voican, I.; Onisai, M.; Nicolescu, A.; Vladareanu, A.N.A.M.; Vladareanu, R. Heparin Induced Thrombocytopenia: A Review. Semin. Thromb. Hemost. 2012, 60, 773–784. [Google Scholar]
- Michel, M.; Lega, J.-C.; Terriou, L. Secondary ITP in adults. La Rev. Med. Interne 2021, 42, 50–57. [Google Scholar] [CrossRef]
- Onisâi, M.; Vlădăreanu, A.-M.; Iordan, I.; Bumbea, H.; Găman, M.; Ciufu, C.; Voican, I.; Cîșleanu, D.; Vasile, D.; Marinescu, C.; et al. Primary, secondary or less frequent causes of immune thrombocytopenia: A case report. Exp. Ther. Med. 2021, 22, 1096. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; González-López, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef] [PubMed]
- Boulware, R.; Refaai, M.A. Why do patients with immune thrombocytopenia (ITP) experience lower bleeding events despite thrombocytopenia? Thromb. Res. 2020, 187, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Lim, W.; Crowther, M.A.; Cohen, A.; Solberg, L. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Bussel, J.B.; Liebman, H.A.; Luning Prak, E.T. The ITP syndrome: Pathogenic and clinical diversity. Blood 2009, 113, 6511–6521. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Mahevas, M.; Bonnotte, B. Immune thrombocytopenia: From pathogenesis to treatment. Rev. Med. Interne 2021, 42, 16–24. [Google Scholar] [CrossRef]
- Audia, S.; Mahévas, M.; Nivet, M.; Ouandji, S.; Ciudad, M.; Bonnotte, B. Immune Thrombocytopenia: Recent Advances in Pathogenesis and Treatments. HemaSphere 2021, 5, e574. [Google Scholar] [CrossRef]
- Liu, X.-G.; Hou, Y.; Hou, M. How we treat primary immune thrombocytopenia in adults. J. Hematol. Oncol. 2023, 16, 4. [Google Scholar] [CrossRef]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Al Mutair, A.; Al Alawi, Z.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J. Pathogenesis in immune thrombocytopenia: New insights. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 306–312. [Google Scholar] [CrossRef]
- Audia, S.; Mahévas, M.; Samson, M.; Godeau, B.; Bonnotte, B. Autoimmunity Reviews Pathogenesis of immune thrombocytopenia. Autoimmun. Rev. 2017, 16, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Roark, J.H.; Bussel, J.B.; Cines, D.B.; Siegel, D.L. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood 2002, 100, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Ballesteros, F.J.; Oregon-Romero, E.; Franco-Topete, R.A.; Govea-Camacho, L.H.; Cruz, A.; Muñoz-Valle, J.F.; Bustos-Rodríguez, F.J.; Pereira-Suárez, A.L.; Palafox-Sánchez, C.A. B-cell activating factor receptor expression is associated with germinal center B-cell maintenance. Exp. Ther. Med. 2019, 17, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12, 611795. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.A.; von Andrian, U.H. How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 2010, 108, 111–165. [Google Scholar] [CrossRef]
- Najaoui, A.; Bakchoul, T.; Stoy, J.; Bein, G.; Rummel, M.J.; Santoso, S.; Sachs, U.J. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur. J. Haematol. 2012, 88, 167–174. [Google Scholar] [CrossRef]
- Amini, S.N.; Nelson, V.S.; Porcelijn, L.; Netelenbos, T.; Zwaginga, J.J.; de Haas, M.; Schipperus, M.R.; Kapur, R. The interplay between GPIb/IX antibodies, platelet hepatic sequestration, and TPO levels in patients with chronic ITP. Blood Adv. 2023, 7, 1066–1069. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, L.; Peng, J.; Hou, M. T cell immune abnormalities in immune thrombocytopenia. J. Hematol. Oncol. 2014, 7, 1–6. [Google Scholar] [CrossRef]
- Yazdanbakhsh, K.; Zhong, H.; Bao, W. Immune dysregulation in immune thrombocytopenia. Semin. Hematol. 2013, 50 (Suppl. S1), S63–S67. [Google Scholar] [CrossRef] [PubMed]
- Swinkels, M.; Rijkers, M.; Voorberg, J.; Vidarsson, G.; Leebeek, F.W.G.; Jansen, A.J.G. Emerging concepts in immune thrombocytopenia. Front. Immunol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Consolini, R.; Legitimo, A.; Caparello, M.C. The Centenary of Immune Thrombocytopenia—Part 1: Revising Nomenclature and Pathogenesis. Front. Pediatr. 2016, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Saitoh, T.; Gotoh, N.; Nitta, Y.; Alkebsi, L.; Kasamatsu, T.; Minato, Y.; Yokohama, A.; Tsukamoto, N.; Handa, H.; et al. The cytokine polymorphisms affecting Th1/Th2 increase the susceptibility to, and severity of, chronic ITP. BMC Immunol. 2017, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Zhao, C.; Li, L.; Peng, J.; Hou, M. CD8+ T cells suppress autologous megakaryocyte apoptosis in idiopathic thrombocytopenic purpura. Br. J. Haematol. 2007, 139, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Ebbo, M.; Audonnet, S.; Grados, A.; Benarous, L.; Mahevas, M.; Godeau, B.; Viallard, J.F.; Piperoglou, C.; Cognet, C.; Farnarier, C.; et al. NK cell compartment in the peripheral blood and spleen in adult patients with primary immune thrombocytopenia. Clin. Immunol. 2017, 177, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Andersson, P.-O.; Jernås, M.; Jacobsson, S.; Carlsson, B.; Carlsson, L.M.S.; Wadenvik, H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat. Med. 2003, 9, 1123–1124. [Google Scholar] [CrossRef]
- Kuter, D.J.; Gernsheimer, T.B. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol. Oncol. Clin. North Am. 2009, 23, 1193–1211. [Google Scholar] [CrossRef]
- Provan, D.; Semple, J.W. Recent advances in the mechanisms and treatment of immune thrombocytopenia. eBioMedicine 2022, 76, 103820. [Google Scholar] [CrossRef]
- Grodzielski, M.; Goette, N.P.; Glembotsky, A.C.; Baroni, M.C.; Méndez-huergo, S.P.; Pierdominici, M.S.; Montero, V.S.; Rabinovich, G.A.; Molinas, F.C.; Heller, P.G.; et al. Multiple concomitant mechanisms contribute to low platelet count in patients with immune thrombocytopenia. Sci. Rep. 2019, 2018, 1–10. [Google Scholar] [CrossRef]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.S.; Jolink, A.T.C.; Amini, S.N.; Zwaginga, J.J.; Netelenbos, T.; Semple, J.W.; Porcelijn, L.; de Haas, M.; Schipperus, M.R.; Kapur, R. Platelets in ITP: Victims in charge of their own fate? Cells 2021, 10, 3235. [Google Scholar] [CrossRef] [PubMed]
- Łukasik, Z.M.; Makowski, M.; Makowska, J.S. From blood coagulation to innate and adaptive immunity: The role of platelets in the physiology and pathology of autoimmune disorders. Rheumatol. Int. 2018, 38, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Scheinberg, P.; Samsel, L.; Rios, O.; Chen, J.; McCoy, J.P.J.; Ghanima, W.; Bussel, J.B.; Young, N.S. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J. Thromb. Haemostasis 2012, 10, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M. Platelet count or bleeding as the outcome in ITP trials? Am. J. Hematol. 2012, 87, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M. Bleeding complications in immune thrombocytopenia. Hematol. Am. Soc. Hematology. Educ. Program 2015, 2015, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Rosthøj, S.; Rajantie, J.; Treutiger, I.; Zeller, B.; Tedgård, U.; Henter, J.-I. Duration and morbidity of chronic immune thrombocytopenic purpura in children: Five-year follow-up of a Nordic cohort. Acta paediatrica 2012, 101, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Praituan, W.; Rojnuckarin, P. Faster platelet recovery by high-dose dexamethasone compared with standard-dose prednisolone in adult immune thrombocytopenia: A prospective randomized trial. J. Thromb. Haemostasis 2009, 7, 1036–1038. [Google Scholar] [CrossRef]
- Cortelazzo, S.; Finazzi, G.; Buelli, M.; Molteni, A.; Viero, P.; Barbui, T. High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood 1991, 77, 31–33. [Google Scholar] [CrossRef]
- Bussel, J.B.; Provan, D.; Shamsi, T.; Cheng, G.; Psaila, B.; Kovaleva, L.; Salama, A.; Jenkins, J.M.; Roychowdhury, D.; Mayer, B.; et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 641–648. [Google Scholar] [CrossRef]
- Kuter, D.J.; Rummel, M.; Boccia, R.; Macik, B.G.; Pabinger, I.; Selleslag, D.; Rodeghiero, F.; Chong, B.H.; Wang, X.; Berger, D.P. Romiplostim or standard of care in patients with immune thrombocytopenia. N. Engl. J. Med. 2010, 363, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Page, L.K.; Psaila, B.; Provan, D.; Michael Hamilton, J.; Jenkins, J.M.; Elish, A.S.; Lesser, M.L.; Bussel, J.B. The immune thrombocytopenic purpura (ITP) bleeding score: Assessment of bleeding in patients with ITP. Br. J. Haematol. 2007, 138, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Michel, M.; Gernsheimer, T.; Ruggeri, M.; Blanchette, V.; Bussel, J.B.; Cines, D.B.; Cooper, N.; Godeau, B.; Greinacher, A.; et al. Standardization of bleeding assessment in immune thrombocytopenia: Report from the International Working Group. Blood 2013, 121, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Gernsheimer, T.; Kuter, D.J. How I treat primary ITP in adult patients who are unresponsive to or dependent on corticosteroid treatment. Blood 2021, 137, 2736–2744. [Google Scholar] [CrossRef]
- Kochhar, M.; Neunert, C. Immune thrombocytopenia: A review of upfront treatment strategies. Blood Rev. 2021, 49, 100822. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Bonnotte, B. Emerging therapies in immune thrombocytopenia. J. Clin. Med. 2021, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Gernsheimer, T.; Stratton, J.; Ballem, P.J.; Slichter, S.J. Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N. Engl. J. Med. 1989, 320, 974–980. [Google Scholar] [CrossRef]
- LeVine, D.N.; Cianciolo, R.E.; Linder, K.E.; Bizikova, P.; Birkenheuer, A.J.; Brooks, M.B.; Salous, A.K.; Nordone, S.K.; Bellinger, D.A.; Marr, H.; et al. Endothelial alterations in a canine model of immune thrombocytopenia. Platelets 2019, 30, 88–97. [Google Scholar] [CrossRef]
- Frederiksen, H.; Ghanima, W. Response of first line treatment with corticosteroids in a population-based cohort of adults with primary immune thrombocytopenia. Eur. J. Intern. Med. 2017, 37, e23–e25. [Google Scholar] [CrossRef]
- Nakazaki, K.; Hosoi, M.; Hangaishi, A.; Ichikawa, M.; Nannya, Y.; Kurokawa, M. Comparison between pulsed high-dose dexamethasone and daily corticosteroid therapy for adult primary immune thrombocytopenia: A retrospective study. Intern. Med. 2012, 51, 859–863. [Google Scholar] [CrossRef]
- Wei, Y.; Ji, X.; Wang, Y.; Wang, J.; Yang, E.; Wang, Z.; Sang, Y.; Bi, Z.; Ren, C.; Zhou, F.; et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: A prospective multicenter randomized trial. Blood 2016, 127, 296–302; quiz 370. [Google Scholar] [CrossRef] [PubMed]
- Mithoowani, S.; Gregory-Miller, K.; Goy, J.; Miller, M.C.; Wang, G.; Noroozi, N.; Kelton, J.G.; Arnold, D.M. High-dose dexamethasone compared with prednisone for previously untreated primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet. Haematol. 2016, 3, e489–e496. [Google Scholar] [CrossRef] [PubMed]
- Onisâi, M.; Vlădăreanu, A.M.; Spînu, A.; Găman, M.; Bumbea, H. Idiopathic thrombocytopenic purpura (ITP)—New era for an old disease. Rom. J. Intern. Med. 2019, 57, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kistangari, G.; McCrae, K.R. Immune thrombocytopenia. Hematol./Oncol. Clin. North Am. 2013, 27, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Kuter, D.J. Antiplatelet Antibody Testing in Immune Thrombocytopenia and Evans Syndrome: Longitudinal Serologic Evolution and Relation to Clinical Features. Blood 2018, 132, 1137. [Google Scholar] [CrossRef]
- Cooper, N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol./Oncol. Clin. North Am. 2009, 23, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Naithani, R.; Kumar, R.; Mahapatra, M.; Tyagi, S.; Saxena, R. Efficacy and safety of anti-D for treatment of adults with immune thrombocytopenia. Platelets 2009, 20, 525–527. [Google Scholar] [CrossRef]
- Lucchini, E.; Zaja, F.; Bussel, J. Rituximab in the treatment of immune thrombocytopenia: What is the role of this agent in 2019? Haematologica 2019, 104, 1124–1135. [Google Scholar] [CrossRef]
- Marangon, M.; Vianelli, N.; Palandri, F.; Mazzucconi, M.G.; Santoro, C.; Barcellini, W.; Fattizzo, B.; Volpetti, S.; Lucchini, E.; Polverelli, N.; et al. Rituximab in immune thrombocytopenia: Gender, age, and response as predictors of long-term response. Eur. J. Haematol. 2017, 98, 371–377. [Google Scholar] [CrossRef]
- Xiao, Z.; Murakhovskaya, I. Rituximab resistance in ITP and beyond. Front. Immunol. 2023, 14, 1215216. [Google Scholar] [CrossRef] [PubMed]
- Vianelli, N.; Auteri, G.; Buccisano, F.; Carrai, V.; Baldacci, E.; Clissa, C.; Bartoletti, D.; Giuffrida, G.; Magro, D.; Rivolti, E.; et al. Refractory primary immune thrombocytopenia (ITP): Current clinical challenges and therapeutic perspectives. Ann. Hematol. 2022, 101, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin receptor agonists: Ten years later. Haematologica 2019, 104, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C.; Lin, N.L. AMG531 stimulates megakaryopoiesis in vitro by binding to Mpl. Cytokine 2004, 25, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Will, B.; Kawahara, M.; Luciano, J.P.; Bruns, I.; Parekh, S.; Erickson-Miller, C.L.; Aivado, M.A.; Verma, A.; Steidl, U. Effect of the nonpeptide thrombopoietin receptor agonist Eltrombopag on bone marrow cells from patients with acute myeloid leukemia and myelodysplastic syndrome. Blood 2009, 114, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Lozano, M.L.; Segú-Vergés, C.; Coma, M.; Álvarez-Roman, M.T.; González-Porras, J.R.; Gutiérrez, L.; Valcárcel, D.; Butta, N. Elucidating the Mechanism of Action of the Attributed Immunomodulatory Role of Eltrombopag in Primary Immune Thrombocytopenia: An In Silico Approach. Int. J. Mol. Sci. 2021, 22, 6907. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Bussel, J.B.; Heck, S.; He, W.; Karpoff, M.; Boulad, N.; Yazdanbakhsh, K. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood 2010, 116, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Schifferli, A.; Kühne, T. Thrombopoietin receptor agonists: A new immune modulatory strategy in immune thrombocytopenia? Semin. Hematol. 2016, 53 (Suppl. S1), S31–S34. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. “Yin-Yang” functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef]
- Markham, A. Avatrombopag: A Review in Thrombocytopenia. Drugs 2021, 81, 1905–1913. [Google Scholar] [CrossRef]
- Ghanima, W.; Geyer, J.T.; Lee, C.S.; Boiocchi, L.; Imahiyerobo, A.A.; Orazi, A.; Bussel, J.B. Bone marrow fibrosis in 66 patients with immune thrombocytopenia treated with thrombopoietin-receptor agonists: A single-center, long-term follow-up. Haematologica 2014, 99, 937–944. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. Optimal use of thrombopoietin receptor agonists in immune thrombocytopenia. Ther. Adv. Hematol. 2019, 10, 2040620719841735. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Arnold, D.M.; McCrae, K.R. Splenectomy for immune thrombocytopenia: Down but not out. Blood 2018, 131, 1172–1182. [Google Scholar] [CrossRef]
- Vianelli, N.; Palandri, F.; Polverelli, N.; Stasi, R.; Joelsson, J.; Johansson, E.; Ruggeri, M.; Zaja, F.; Cantoni, S.; Catucci, A.E.; et al. Splenectomy as a curative treatment for immune thrombocytopenia: A retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica 2013, 98, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.; Álvarez-Román, M.T.; Monzón-Manzano, E.; Acuña, P.; Arias-Salgado, E.G.; Rivas-Pollmar, I.; Martín-Salces, M.; de Miguel, B.; Martínez Montalbán, E.; Jiménez-Yuste, V.; et al. Study of platelet kinetics in immune thrombocytopenia to predict splenectomy response. Br. J. Haematol. 2024, 204, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Yi, H.G.; Kim, C.S.; Hong, J.; Park, J.; Lee, J.H.; Kim, H.Y.; Kim, H.J.; Zang, D.Y.; Kim, S.H.; et al. Clinical Outcome and Predictive Factors in the Response to Splenectomy in Elderly Patients with Primary Immune Thrombocytopenia: A Multicenter Retrospective Study. Acta Haematol. 2016, 135, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Onisâi, M.; Vlădăreanu, A.-M.; Nica, A.; Spînu, A.; Găman, M.; Bumbea, H.; Voican, I.; Iordan, I.; Alexandru, A.; Zdrenghea, M.; et al. Splenectomy in Lymphoproliferative Disorders: A Single Eastern European Center Experience. Medicina 2019, 56, 12. [Google Scholar] [CrossRef] [PubMed]
- Fitzer-Attas, C.J.; Lowry, M.; Crowley, M.T.; Finn, A.J.; Meng, F.; DeFranco, A.L.; Lowell, C.A. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp. Med. 2000, 191, 669–682. [Google Scholar] [CrossRef]
- Connell, N.T.; Berliner, N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood 2019, 133, 2027–2030. [Google Scholar] [CrossRef]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef]
- Duan, R.; Goldmann, L.; Brandl, R.; Spannagl, M.; Weber, C.; Siess, W.; von Hundelshausen, P. Effects of the Btk-Inhibitors Remibrutinib (LOU064) and Rilzabrutinib (PRN1008) with Varying Btk Selectivity over Tec on Platelet Aggregation and in vitro Bleeding Time. Front. Cardiovasc. Med. 2021, 8, 749022. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.C.; McDonald, V. FcRn antagonists in ITP. Ann. Blood 2020, 6. [Google Scholar] [CrossRef]
- Castelli, R.; Gidaro, A.; Deliliers, G.L. Risk of Thrombosis In Elderly Immune Primary Trombocytopenic Patients Treated with Thrombopoietin Receptors Agonists. J. Thromb. Thrombolysis 2020, 50, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Broome, C.M.; Röth, A.; Kuter, D.J.; Scully, M.; Smith, R.; Wang, J.; Reuter, C.; Hobbs, W.; Daak, A. Safety and efficacy of classical complement pathway inhibition with sutimlimab in chronic immune thrombocytopenia. Blood Adv. 2023, 7, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Wu, Y.; Zhou, H.; Qin, P.; Ni, H.; Peng, J.; Hou, M. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets 2015, 26, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Kapur, R.; Semple, J.W. Platelets as immune-sensing cells. Blood Adv. 2016, 1, 10–14. [Google Scholar] [CrossRef]
- Rong, W.; Zhang, Y.X.; Xu, S.S.; Shi, J.M. Lymphocyte subsets in primary immune thrombocytopenia. Blood Coagul. Fibrinolysis 2014, 25, 816–819. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Li, D.Q.; Wang, J.; Hou, Y.; Sun, L.; Peng, J.; Hou, M. Effect and mechanism of low-dose chidamide on the treatment of primary immune thrombocytopenia. Chin. J. Hematol. 2020, 41, 292–296. [Google Scholar] [CrossRef]
- Zhou, H.; Qin, P.; Liu, Q.; Yuan, C.; Hao, Y.; Zhang, H.; Wang, Z.; Ran, X.; Chu, X.; Yu, W.; et al. A prospective, multicenter study of low dose decitabine in adult patients with refractory immune thrombocytopenia. Am. J. Hematol. 2019, 94, 1374–1381. [Google Scholar] [CrossRef]
- Han, P.; Hou, Y.; Zhao, Y.; Liu, Y.; Yu, T.; Sun, Y.; Wang, H.; Xu, P.; Li, G.; Sun, T.; et al. Low-dose decitabine modulates T-cell homeostasis and restores immune tolerance in immune thrombocytopenia. Blood 2021, 138, 674–688. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. Relative potency of the thrombopoietin receptor agonists eltrombopag, avatrombopag and romiplostim in a patient with chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 168. [Google Scholar] [CrossRef]
- Nomoto, H.; Morimoto, N.; Miura, K.; Watanabe, S.; Takaoka, Y.; Maeda, H.; Sasaki, T.; Koyashiki, Y.; Kurata, H.; Numao, N.; et al. Lusutrombopag is effective and safe in patients with chronic liver disease and severe thrombocytopenia: A multicenter retrospective study. BMC Gastroenterol. 2020, 20, 427. [Google Scholar] [CrossRef]
- Liu, X.; Bai, Y.; Wang, T.; Song, Y.; Sun, F.; Xia, R.; Zhu, F.; Ma, J.; Lu, Q.; Ye, X.; et al. Recombinant human thrombopoietin (rhTPO) of different dosing regimens for refractory/relapsed primary immune thrombocytopenia: A multicenter, randomized controlled trial and pharmacokinetics study. Platelets 2023, 34, 2157806. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapy | Mechanism of Action | Drug and Dosage | Durability of Effect | Side Effects and Cautions |
|---|---|---|---|---|
| Steroids | Broad action on immune cells (macrophages, B and T cells), limiting platelet destruction | Steroid Prednisone/Prednisolone 1–2 mg orally for 1–2 weeks, followed by gradual tapering | Response rate in 60–80% of patients with sustained response after discontinuation in 30–50% of patients | Weight gain, insomnia, acne, mood changes, cushingoid appearance, glucose intolerance, osteoporosis, risk of fracture, gastrointestinal symptoms, neuropsychiatric symptoms |
| Dexamethasone 20–40 mg for 4 days every 2–4 weeks; maximum of 4 cycles | ||||
| Intravenous Immunoglobulin | Reduction in platelet destruction by inhibiting splenic macrophages Enhancement of antiplatelet antibodies clearance by FcRn saturation | Intravenous Immunoglobulin 1–2 g/kg for 4–5 days | Transient response lasting 1–4 weeks in about 80% of patients | Headache, aseptic meningitis, renal failure |
| Thrombopoietin receptor agonists | Megakaryocyte-induced increase in platelet production | Eltrombopag 1–10 μg per kilogram, subcutaneously once a week | Response achieved in 1–2 weeks and maintained in 40–60% of patients continuing therapy | Headache, muscle pain, possible increased risk of thrombosis and myelofibrosis |
| Romiplostim 25–75 mg orally daily | Gastrointestinal symptoms, hepatocytolysis, cataract, possible increased risk of thrombosis and myelofibrosis—should be taken 4 hr after and 2 hr before food containing cations | |||
| Avatrombopag 5–40 mg orally daily | Response is maintained in 10–30% of patients Response achieved in 1–2 weeks in 65% of patients | Headache, arthralgia, possible increased risk of thrombosis | ||
| Lusutrombopag 3 mg orally daily for 7 days—approved in patients with chronic liver disease who are scheduled for an invasive procedure | 78% of patients did not require platelet transfusion prior to the invasive procedure | Increased risk of thrombosis, headache | ||
| Hetrombopag Olamine 2.-7.5 mg orally daily (approved only in China) | Response achieved in 6 days, with a peak at 12–14 days | Hepatocytolysis, hyperuricemia, acute myocardial infarction, anemia | ||
| rhTPO subcutaneously (approved only in China) | Significant increase in platelet count in 7 days | Fever, rash, dizziness, pain at injection site, high blood pressure | ||
| Immunosuppressants | Inhibition of T and B cells | Azathioprine 1–2 mg per kilogram orally (maximum 150 mg daily) | Response rate in 30–60% of patients in 1–2 weeks | Weakness, sweating, neutropenia, increased liver function, increased risk of cancer |
| Mycophenolate mofetil 500 mg orally twice daily for 2 weeks with gradual increase to a maximum of 1 g twice daily | Response rate in 30–60% of patients in 4–8 weeks | Headache, gastrointestinal symptoms, fungal skin infections, increased risk of cancer | ||
| Cyclophosphamide pulse therapy 1–1.5 g/m2 intravenously | Response rate in 85% of patients; mean time to response 7 weeks | Neutropenia, infections, deep venous thrombosis | ||
| Danazol 400–800 orally daily | Response rate in 30–60% of patients within 3–6 months | Hirsutism, acne, amenorrhea, hepatic dysfunction; contraindicated in prostatic cancer | ||
| Dapsone 75–100 mg orally daily | Response rate in 30–60% patients in 3 weeks | Gastrointestinal symptoms, methemoglobinemia, rash, hemolytic anemia (in patients with glucose-6-phosphate dehydrogenase deficiency) | ||
| Syk inhibitors | Reduction in platelet destruction by inhibiting macrophage phagocytosis | Fostamatinib 50–150 mg orally twice daily | Response achieved and maintained in 18–43% of patients | Hypertension, nausea, diarrhea, increased liver enzymes |
| BTK inhibitors | Reduction in platelet destruction by inhibiting macrophage phagocytosis | Rilzabrutinib 400 mg twice daily | Phase 3 Trial (NCT04562766) Response in 40% of patients who received 400 mg twice a day; median time to response 11.5 days | Diarrhea, nausea, fatigue; no grade 3–4 side effects |
| FcRn blockers | Enhancement of antiplatelet antibody clearance by decreasing peripheral platelet destruction and immune response against megakaryocytes | Rozanolixizumab single-dose 15–21 mg/kg subcutaneous infusion or fractioned doses | Phase 3 Trial (NCT00718692) Better response in single-dose regimen; response in 66.7% patients; median time to response 5–7 days | Headache, diarrhea, pyrexia, nausea, infections |
| Nipocalimab Intravenous, subcutaneous | Phase 2 Trial (AIHA, NCT04119050) | |||
| Efgartigimod 10 mg/kg weekly for 4 weeks, then weekly or every 2 weeks depending on response | Phase 3 Trial (NCT04188379) Response in 51.2% of patients; early platelet count increase | Bruising, headache, hematuria, petechiae | ||
| Recombinant immunoglobulin multimers | Reduction in platelet destruction by inhibiting splenic of macrophage phagocytosis Enhancement of antiplatelet antibody clearance by saturation of FcRn | GL-2045 CSL730 M254 | Phase 1 Trial | |
| Complement inhibitors | Decrease complement-dependent cytotoxicity | Sutimlimab (C1s inhibitor) 6.5 g if <75 kg or 7.5 g if ≥75 kg biweekly by intravenous infusion | Phase 2 Trial (NCT04669600) Response obtained in 42% of patients. The median time to first platelet response after the first dose was 22 days | Headache, fatigue |
| Inhibition of platelet desialylation | Neuramidase-1 inhibitor | Oseltamivir 75 mg twice a day for 5 days | Phase 1/2 Trial (NCT06520049) Higher response rate in patients receiving Oseltamivir and Dexamethasone (86%) than Dexamethasone alone (53%) | |
| B-cell-depleting therapies—anti-CD20 antibodies | Decrease in short-lived plasma cells by reducing their B-cell precursors, thus decreasing antiplatelet antibodies Restoration of T-cell tolerance | Rituximab 375 mg per square meter of body surface area intravenously weekly for 4 wk (off label use) | Sustained response in 60% of patients at 6 months and 30% at 2 yr; treatment can be repeated | Infusion-related side effects: chills, bronchospasm, neutropenia, hypogammaglobulinemia, serum sickness, increased risk of infections and progressive multifocal leukoencephalopathy; caution in patients with HBV infection or previous HBV infection |
| Veltuzumab 80–320 mg 2 doses administered two weeks apart, initially intravenously, then subcutaneously (off label use) | Overall response rate 55%. Median time from first dose to response onset 23 days | Hypersensitivity reaction, fever, body aches, nausea | ||
| Obinutuzumab 1000 mg intravenously at days 1, 8, and 15 | Remission rate 100%; only one of four patients relapsed during a follow-up of 15 months | Peripheral neuropathy, fatigue, lymphopenia | ||
| Plasma cell-targeting therapies | Decrease in short- and long-lived plasma cells and reducing the antiplatelet antibodies | Proteasome inhibitors: Bortezomib 1.3 mg/m2 subcutaneously or intravenously on days 1, 4, and 8 every 3 weeks for a total of 3 cycles | Response rate of 100%; 37.5% complete response (Bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 for 2 cycles) | Peripheral neuropathy, fatigue, lymphopenia |
| Anti-CD38 monoclonal antibodies: Daratumumab subcutaneously | Phase II study with safety run-in: 3 patients received 4 weekly subcutaneous daratumumab—2 patients responded, 1 relapsed | Bronchospasm, hypoxia, dyspnea, hypertension, headache | ||
| Mezagitamab Subcutaneously | Phase 2 trial (NCT042789) no results posted | Headache, dizziness, chills, mild injection site reactions | ||
| B-cell- and plasma cell-targeting therapies | Decrease in short- and long-lived plasma cells, thus decreasing antiplatelet antibodies | Anti-CD19 - Inebilizumab - Obexelimab | No clinical trials for ITP | |
| T-cell-targeting therapies | Inhibition of effector T cells Inhibition of follicular helper T cells | Anti-CD154 Rupulizumab (hu5c8) 20 mg/kg once every 4 weeks for 12 weeks | Response in 43% of patients | Thromboembolic events |
| Toralizumab (IDEC-131) 5–20 mg/kg | Response in 16% of patients | Thromboembolic events | ||
| Letolizumab (BMS-986004) 75–1500 mg intravenously | Response in 20–40% of patients | Anemia, hemorrhage, fatigue, headache, body ache | ||
| Anti-CD40 BI 655064 | No available results from clinical trials for ITP | Infection, opportunistic infections, neutropenia, lymphopenia, alopecia | ||
| Restoration of regulatory | IL-21 inhibitors | No clinical trials for ITP | ||
| Low-dose IL-2 1 million IU/day 5 days per week for 2–4 weeks | Phase 2 Trial (NCT01988506) Improvement in platelet count in 2 of 3 patients | Injection site reaction0061 | ||
| Epigenetic modulation | Chidamide (Tucidinostat) 2.5 or 5 mg orally, twice a week for four weeks, one cycle | Phase 2 Trial (NCT03838354) | Diarrhea, fatigue, nausea | |
| Low-dose decitabine 3.5 mg/m2 IV for 3 consecutive days, repeated every 28 days for three cycles | Complete response in 17.78%; partial response in 33.33%. Median time to response 28 days | Nausea, fever; no grade 3–4 adverse events | ||
| Splenectomy | Reduction in platelet destruction by inhibiting splenic macrophage phagocytosis Removal of the maintenance site of the autoimmune response | Favorable response obtained in 89% of patients | Postoperative complications, major bleeding higher risk for the elderly group, infections, thrombotic events |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mititelu, A.; Onisâi, M.-C.; Roșca, A.; Vlădăreanu, A.M. Current Understanding of Immune Thrombocytopenia: A Review of Pathogenesis and Treatment Options. Int. J. Mol. Sci. 2024, 25, 2163. https://doi.org/10.3390/ijms25042163
Mititelu A, Onisâi M-C, Roșca A, Vlădăreanu AM. Current Understanding of Immune Thrombocytopenia: A Review of Pathogenesis and Treatment Options. International Journal of Molecular Sciences. 2024; 25(4):2163. https://doi.org/10.3390/ijms25042163
Chicago/Turabian StyleMititelu, Alina, Minodora-Cezarina Onisâi, Adrian Roșca, and Ana Maria Vlădăreanu. 2024. "Current Understanding of Immune Thrombocytopenia: A Review of Pathogenesis and Treatment Options" International Journal of Molecular Sciences 25, no. 4: 2163. https://doi.org/10.3390/ijms25042163