
Mitochondrial Quantity and Quality in Age-Related Sarcopenia
 , , ,
, , , 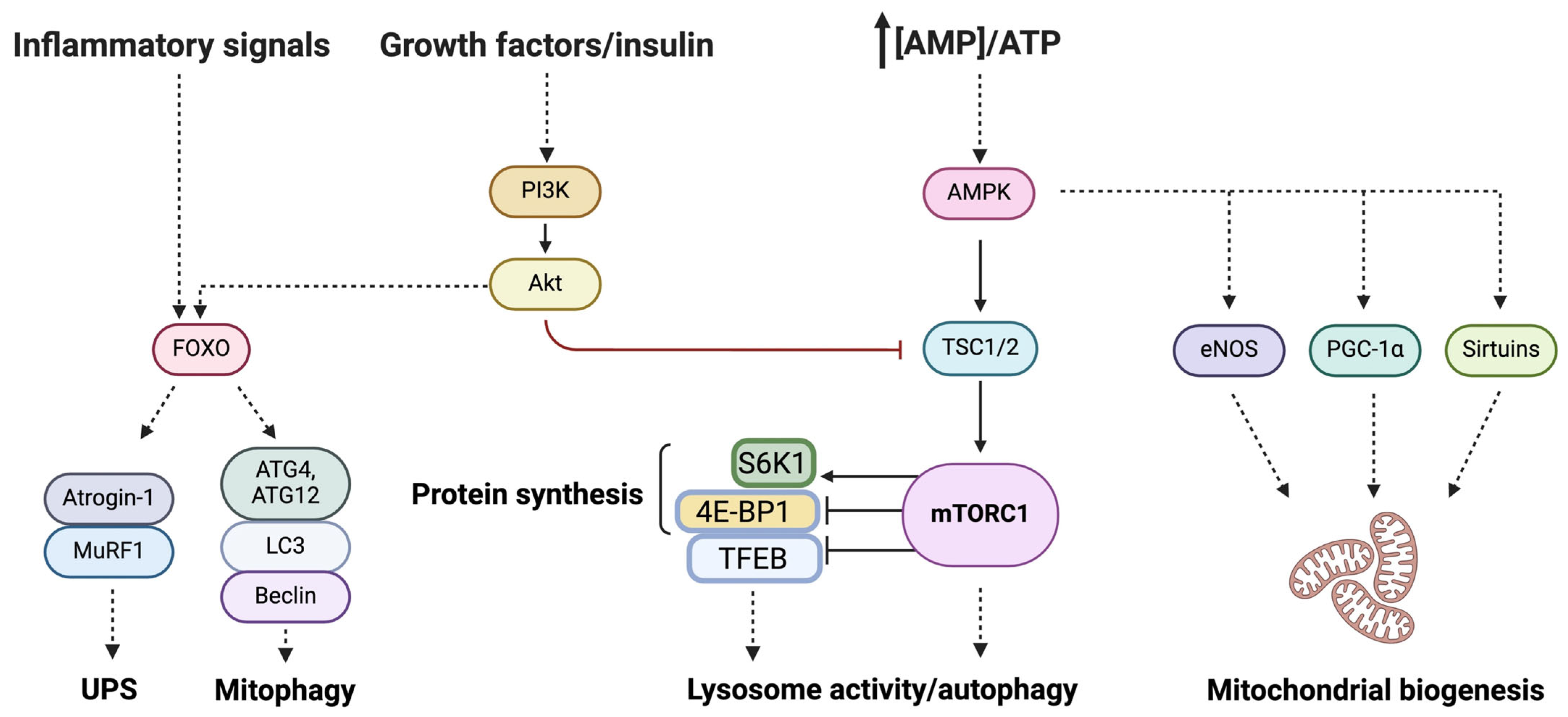{kind=link}
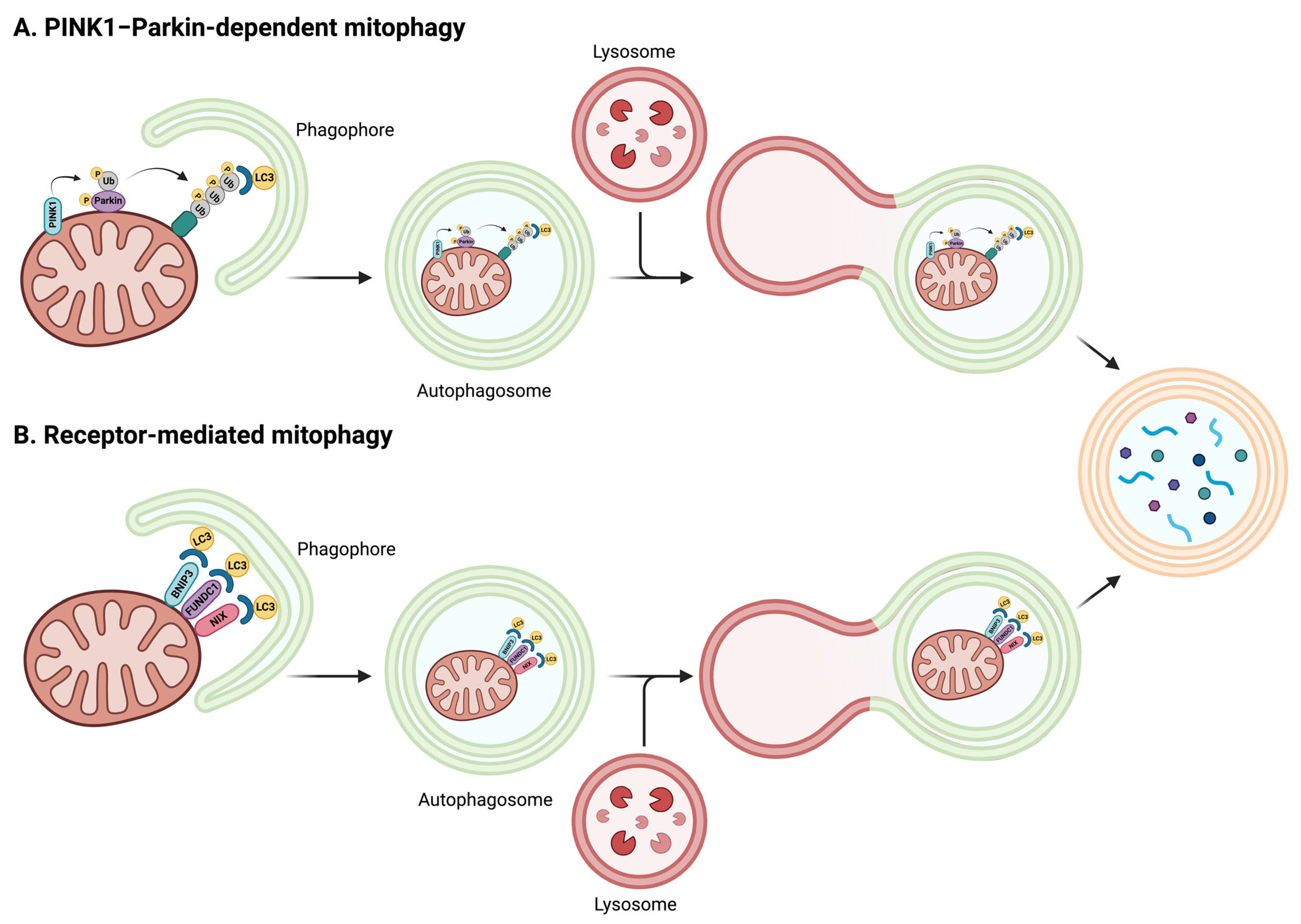{kind=link}
Abstract
:1. Introduction
2. Age-Related Changes in Muscle and Mitochondrial Function
2.1. Age-Related Changes in Muscle Mass, Strength, and Function
2.2. Mitochondria and Muscle Aging
3. Bridging Energy Production to Organelle Quality: The Role of Mitophagy in Muscle (Patho)physiology
4. Mitochondrial Function Recovery: From Energizing Mitochondria to Organelle’s Transplantation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janssen, I.; Heymsfield, S.B.; Wang, Z.M.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. (1985) 2000, 89, 81–88. [Google Scholar] [CrossRef]
- Swalsingh, G.; Pani, P.; Bal, N.C. Structural functionality of skeletal muscle mitochondria and its correlation with metabolic diseases. Clin. Sci. 2022, 136, 1851–1871. [Google Scholar] [CrossRef]
- Coelho-Junior, H.J.; Picca, A.; Calvani, R.; Uchida, M.C.; Marzetti, E. If my muscle could talk: Myokines as a biomarker of frailty. Exp. Gerontol. 2019, 127, 110715. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Coenen-Schimke, J.M.; Rys, P.; Nair, K.S. Changes in myosin heavy chain mRNA and protein expression in human skeletal muscle with age and endurance exercise training. J. Appl. Physiol. (1985) 2005, 99, 95–102. [Google Scholar] [CrossRef]
- Verdijk, L.B.; Snijders, T.; Drost, M.; Delhaas, T.; Kadi, F.; Van Loon, L.J.C. Satellite cells in human skeletal muscle; From birth to old age. Age 2014, 36, 545–557. [Google Scholar] [CrossRef]
- Ogata, T.; Yamasaki, Y. Scanning electron-microscopic studies on the three-dimensional structure of mitochondria in the mammalian red, white and intermediate muscle fibers. Cell Tissue Res. 1985, 241, 251–256. [Google Scholar] [CrossRef]
- Hoppeler, H. Exercise-induced ultrastructural changes in skeletal muscle. Int. J. Sports Med. 1986, 7, 187–204. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; De Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 2023, 5, 2047–2061. [Google Scholar] [CrossRef]
- Sirago, G.; Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Marzetti, E. Mammalian target of rapamycin (mTOR) signaling at the crossroad of muscle fiber fate in sarcopenia. Int. J. Mol. Sci. 2022, 23, 13823. [Google Scholar] [CrossRef]
- Beaudart, C.; Zaaria, M.; Pasleau, F.; Reginster, J.-Y.; Bruyère, O. Health outcomes of sarcopenia: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0169548. [Google Scholar] [CrossRef]
- Veronese, N.; Demurtas, J.; Soysal, P.; Smith, L.; Torbahn, G.; Schoene, D.; Schwingshackl, L.; Sieber, C.; Bauer, J.; Cesari, M.; et al. Sarcopenia and health-related outcomes: An umbrella review of observational studies. Eur. Geriatr. Med. 2019, 10, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R.; Dunne, R.F.; Giri, S.; Shachar, S.S.; Caan, B.J. Sarcopenia in the older adult with cancer. J. Clin. Oncol. 2021, 39, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Colloca, G.; Di Capua, B.; Bellieni, A.; Cesari, M.; Marzetti, E.; Valentini, V.; Calvani, R. Muscoloskeletal aging, sarcopenia and cancer. J. Geriatr. Oncol. 2019, 10, 504–509. [Google Scholar] [CrossRef]
- Mesinovic, J.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sarcopenia and type 2 diabetes mellitus: A bidirectional relationship. Diabetes Metab. Syndr. Obes. 2019, 12, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.; Springer, J.I.; Anker, S.D. Muscle wasting and sarcopenia in heart failure and beyond: Update 2017. ESC Heart Fail. 2017, 4, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Landi, F.; Liperoti, R.; Fusco, D.; Mastropaolo, S.; Quattrociocchi, D.; Proia, A.; Tosato, M.; Bernabei, R.; Onder, G. Sarcopenia and mortality among older nursing home residents. J. Am. Med. Dir. Assoc. 2012, 13, 121–126. [Google Scholar] [CrossRef]
- Landi, F.; Cruz-Jentoft, A.J.; Liperoti, R.; Russo, A.; Giovannini, S.; Tosato, M.; Capoluongo, E.; Bernabei, R.; Onder, G. Sarcopenia and mortality risk in frail older persons aged 80 years and older: Results from IlSIRENTE study. Age Ageing 2013, 42, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, D.L.; Landi, F.; Volpato, S.; Corsonello, A.; Meloni, E.; Bernabei, R.; Onder, G. Association of sarcopenia with short- and long-term mortality in older adults admitted to acute care wards: Results from the CRIME study. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Kyle, U.G.; Genton, L.; Hans, D.; Karsegard, L.; Slosman, D.O.; Pichard, C. Age-related differences in fat-free mass, skeletal muscle, body cell mass and fat mass between 18 and 94 years. Eur. J. Clin. Nutr. 2001, 55, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; A quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef]
- Gallagher, D.; Visser, M.; De Meersman, R.E.; Sepúlveda, D.; Baumgartner, R.N.; Pierson, R.N.; Harris, T.; Heymsfield, S.B. Appendicular skeletal muscle mass: Effects of age, gender, and ethnicity. J. Appl. Physiol. (1985) 1997, 83, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Kallman, D.A.; Plato, C.C.; Tobin, J.D. The Role of muscle loss in the age-related decline of grip strength: Cross-Sectional and longitudinal perspectives. J. Gerontol. 1990, 45, M82–M88. [Google Scholar] [CrossRef]
- Metter, E.J.; Conwit, R.; Tobin, J.; Fozard, J.L. Age-associated loss of power and strength in the upper extremities in women and men. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52A, B267–B276. [Google Scholar] [CrossRef]
- Vandervoort, A.A.; McComas, A.J. Contractile changes in opposing muscles of the human ankle joint with aging. J. Appl. Physiol. (1985) 1986, 61, 361–367. [Google Scholar] [CrossRef]
- Frontera, W.R.; Hughes, V.A.; Fielding, R.A.; Fiatarone, M.A.; Evans, W.J.; Roubenoff, R. Aging of skeletal muscle: A 12-yr longitudinal study. J. Appl. Physiol. (1985) 2000, 88, 1321–1326. [Google Scholar] [CrossRef]
- Winegard, K.J.; Hicks, A.L.; Sale, D.G.; Vandervoort, A.A. A 12-year follow-up study of ankle muscle function in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51A, B202–B207. [Google Scholar] [CrossRef]
- Coelho-Junior, H.J.; Marzetti, E.; Picca, A.; Tosato, M.; Calvani, R.; Landi, F. Sex- and age-specific normative values of lower extremity muscle power in italian community-dwellers. J. Cachexia Sarcopenia Muscle 2024, 15, 45–54. [Google Scholar] [CrossRef]
- Simoneau, E.M.; Billot, M.; Martin, A.; Van Hoecke, J. Antagonist mechanical contribution to resultant maximal torque at the ankle joint in young and older men. J. Electromyogr. Kinesiol. 2009, 19, e123–e131. [Google Scholar] [CrossRef]
- Clark, B.C.; Manini, T.M. Sarcopenia =/= dynapenia. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 829–834. [Google Scholar] [CrossRef]
- Vandervoort, A.A. Aging of the human neuromuscular system. Muscle Nerve 2002, 25, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.F.; Kiely, D.K.; Herman, S.; Leveille, S.G.; Mizer, K.; Frontera, W.R.; Fielding, R.A. The relationship between leg power and physical performance in mobility-limited older people. J. Am. Geriatr. Soc. 2002, 50, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.F.; Leveille, S.G.; Kiely, D.K.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. A comparison of leg power and leg strength within the InCHIANTI study: Which influences mobility more? J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 728–733. [Google Scholar] [CrossRef]
- Byrne, C.; Faure, C.; Keene, D.J.; Lamb, S.E. Ageing, muscle power and physical function: A systematic review and implications for pragmatic training interventions. Sports Med. 2016, 46, 1311–1332. [Google Scholar] [CrossRef] [PubMed]
- Rantanen, T.; Avela, J. Leg extension power and walking speed in very old people living independently. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52A, M225–M231. [Google Scholar] [CrossRef]
- Strandkvist, V.; Larsson, A.; Pauelsen, M.; Nyberg, L.; Vikman, I.; Lindberg, A.; Gustafsson, T.; Röijezon, U. Hand grip strength is strongly associated with lower limb strength but only weakly with postural control in community-dwelling older adults. Arch. Gerontol. Geriatr. 2021, 94, 104345. [Google Scholar] [CrossRef]
- Tatangelo, T.; Muollo, V.; Ghiotto, L.; Schena, F.; Rossi, A.P. Exploring the association between handgrip, lower limb muscle strength, and physical function in older adults: A narrative review. Exp. Gerontol. 2022, 167, 111902. [Google Scholar] [CrossRef]
- Jakobi, J.M.; Rice, C.L. Voluntary muscle activation varies with age and muscle group. J. Appl. Physiol. (1985) 2002, 93, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.I.; Thom, J.M.; Davis, M.G.; Fox, K.R.; Birch, K.M.; Narici, M.V. Reduced plantarflexor specific torque in the elderly is associated with a lower activation capacity. Eur. J. Appl. Physiol. 2004, 92, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Stackhouse, S.K.; Stevens, J.E.; Lee, S.C.; Pearce, K.M.; Snyder-Mackler, L.; Binder-Macleod, S.A. Maximum voluntary activation in nonfatigued and fatigued muscle of young and elderly individuals. Phys. Ther. 2001, 81, 1102–1109. [Google Scholar] [CrossRef]
- Yue, G.H.; Ranganathan, V.K.; Siemionow, V.; Liu, J.Z.; Sahgal, V. Older adults exhibit a reduced ability to fully activate their biceps brachii muscle. J. Gerontol. A Biol. Sci. Med. Sci. 1999, 54, M249–M253. [Google Scholar] [CrossRef] [PubMed]
- Hortobágyi, T.; Del Olmo, M.F.; Rothwell, J.C. Age reduces cortical reciprocal inhibition in humans. Exp. Brain Res. 2006, 171, 322–329. [Google Scholar] [CrossRef]
- Kido, A.; Tanaka, N.; Stein, R.B. Spinal excitation and inhibition decrease as humans age. Can. J. Physiol. Pharmacol. 2004, 82, 238–248. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C.; Sjöström, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Narici, M.V.; Maganaris, C.N. Adaptability of elderly human muscles and tendons to increased loading. J. Anat. 2006, 208, 433–443. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Marini, F.; Landi, F.; Marzetti, E. Circulating inflammatory, mitochondrial dysfunction, and senescence-related markers in older adults with physical frailty and sarcopenia: A BIOSPHERE exploratory study. Int. J. Mol. Sci. 2022, 23, 14006. [Google Scholar] [CrossRef]
- Picca, A.; Coelho-Junior, H.J.; Calvani, R.; Marzetti, E.; Vetrano, D.L. Biomarkers shared by frailty and sarcopenia in older adults: A systematic review and meta-analysis. Ageing Res. Rev. 2022, 73, 101530. [Google Scholar] [CrossRef]
- Marzetti, E.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Primiano, A.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; et al. Circulating mitochondrial-derived vesicles, inflammatory biomarkers and amino acids in older adults with physical frailty and sarcopenia: A preliminary BIOSPHERE multi-marker study using sequential and orthogonalized covariance selection—linear discriminant analysis. Front. Cell Dev. Biol. 2020, 8, 564417. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Gaitanos, G.C.; Williams, C.; Boobis, L.H.; Brooks, S. Human muscle metabolism during intermittent maximal exercise. J. Appl. Physiol. (1985) 1993, 75, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Scott, W.; Stevens, J.; Binder-Macleod, S.A. Human skeletal muscle fiber type classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Joseph, A.M.; Saleem, A.; Uguccioni, G.; Collu-Marchese, M.; Lai, R.Y.J.; Nguyen, L.M.D.; Hood, D.A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochim. Biophys. Acta 2010, 1800, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.E.; Karunadharma, P.P.; Basisty, N.; Johnson, R.; Beyer, R.P.; Maccoss, M.J.; Rabinovitch, P.S.; Marcinek, D.J. Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner. Aging Cell 2016, 15, 89–99. [Google Scholar] [CrossRef]
- McDonagh, B.; Sakellariou, G.K.; Smith, N.T.; Brownridge, P.; Jackson, M.J. Differential cysteine labeling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J. Proteome Res. 2014, 13, 5008–5021. [Google Scholar] [CrossRef]
- Welle, S.; Bhatt, K.; Shah, B.; Needler, N.; Delehanty, J.M.; Thornton, C.A. Reduced amount of mitochondrial dna in aged human muscle. J. Appl. Physiol. (1985) 2003, 94, 1479–1484. [Google Scholar] [CrossRef]
- Huang, J.H.; Joseph, A.M.; Ljubicic, V.; Iqbal, S.; Hood, D.A. Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 138–146. [Google Scholar] [CrossRef]
- Ljubicic, V.; Joseph, A.-M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging 2009, 1, 818–830. [Google Scholar] [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 119–128. [Google Scholar] [CrossRef]
- Joseph, A.-M.; Ljubicic, V.; Adhihetty, P.J.; Hood, D.A. Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am. J. Physiol. Cell Physiol. 2010, 298, C1308–C1314. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.-M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.-D.M.-D.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef]
- Southern, W.M.; Nichenko, A.S.; Tehrani, K.F.; McGranahan, M.J.; Krishnan, L.; Qualls, A.E.; Jenkins, N.T.; Mortensen, L.J.; Yin, H.; Yin, A.; et al. PGC-1α overexpression partially rescues impaired oxidative and contractile pathophysiology following volumetric muscle loss injury. Sci. Rep. 2019, 9, 4079. [Google Scholar] [CrossRef] [PubMed]
- Leick, L.; Lyngby, S.S.; Wojtasewski, J.F.P.; Pilegaard, H. PGC-1alpha is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp. Gerontol. 2010, 45, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, C.M.A.; Virbasius, J.V.; Scarpulla, R.C. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 1993, 7, 2431–2445. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Knutti, D.; Brogli, K.; Uhlmann, T.; Kralli, A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J. Biol. Chem. 2003, 278, 9013–9018. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor a regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Fisher, R.P.; Parisi, M.A.; Clayton, D.A. Flexible recognition of rapidly evolving promoter sequences by mitochondrial transcription factor 1. Genes Dev. 1989, 3, 2202–2217. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.V.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.-M.; Leeuwenburgh, C.; Lezza, A.M.S. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim. Biophys. Acta 2014, 1840, 2184–2191. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Fracasso, F.; Marzetti, E.; Calvani, R.; Leeuwenburgh, C.; Russo, F.; Lezza, A.M.S.; Pesce, V. Differences in liver TFAM binding to mtDNA and mtDNA damage between aged and extremely aged rats. Int. J. Mol. Sci. 2019, 20, 2601. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R.; et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef]
- Ringholm, S.; Biensø, R.S.; Kiilerich, K.; Guadalupe-Grau, A.; Aachmann-Andersen, N.J.; Saltin, B.; Plomgaard, P.; Lundby, C.; Wojtaszewski, J.F.P.; Calbet, J.A.; et al. Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E649–E658. [Google Scholar] [CrossRef]
- Geng, T.; Li, P.; Okutsu, M.; Yin, X.; Kwek, J.; Zhang, M.; Yan, Z. PGC-1α Plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2010, 298, C572–C579. [Google Scholar] [CrossRef]
- Grevendonk, L.; Connell, N.J.; McCrum, C.; Fealy, C.E.; Bilet, L.; Bruls, Y.M.H.; Mevenkamp, J.; Schrauwen-Hinderling, V.B.; Jörgensen, J.A.; Moonen-Kornips, E.; et al. Impact of aging and exercise on skeletal muscle mitochondrial capacity, energy metabolism, and physical function. Nat. Commun. 2021, 12, 4773. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Saini, S.K.; Calvani, R.; Landi, F.; Bernabei, R.; et al. Advanced age is associated with iron dyshomeostasis and mitochondrial DNA damage in human skeletal muscle. Cells 2019, 8, 1525. [Google Scholar] [CrossRef]
- Picca, A.; Saini, S.K.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Wohlgemuth, S.E.; Xiao, R.; Calvani, R.; et al. Altered expression of mitoferrin and frataxin, larger labile iron pool and greater mitochondrial DNA damage in the skeletal muscle of older adults. Cells 2020, 9, 2579. [Google Scholar] [CrossRef]
- de Jong, J.C.B.C.; Attema, B.J.; van der Hoek, M.D.; Verschuren, L.; Caspers, M.P.M.; Kleemann, R.; van der Leij, F.R.; van den Hoek, A.M.; Nieuwenhuizen, A.G.; Keijer, J. Sex differences in skeletal muscle-aging trajectory: Same processes, but with a different ranking. Geroscience 2023, 45, 2367–2386. [Google Scholar] [CrossRef]
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell 2019, 18, e12943. [Google Scholar] [CrossRef]
- Joseph, G.A.; Wang, S.X.; Jacobs, C.E.; Zhou, W.; Kimble, G.C.; Tse, H.W.; Eash, J.K.; Shavlakadze, T.; Glass, D.J. Partial inhibition of mTORC1 in aged rats counteracts the decline in muscle mass and reverses molecular signaling associated with sarcopenia. Mol. Cell Biol. 2019, 39, e00141. [Google Scholar] [CrossRef]
- Xie, W.Q.; Xiao, W.F.; Tang, K.; Wu, Y.X.; Hu, P.W.; Li, Y.S.; Duan, Y.; Lv, S. Caloric restriction: Implications for sarcopenia and potential mechanisms. Aging 2020, 12, 24441–24452. [Google Scholar] [CrossRef]
- Picca, A.; Triolo, M.; Wohlgemuth, S.E.; Martenson, M.S.; Mankowski, R.T.; Anton, S.D.; Marzetti, E.; Leeuwenburgh, C.; Hood, D.A. Relationship between mitochondrial quality control markers, lower extremity tissue composition, and physical performance in physically inactive older adults. Cells 2023, 12, 183. [Google Scholar] [CrossRef]
- Pedersen, L.; Hojman, P. Muscle-to-organ cross talk mediated by myokines. Adipocyte 2012, 1, 164–167. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, J.L.; He, L.; Virecoulon Giudici, K.; Guyonnet, S.; Parini, A.; Dray, C.; Valet, P.; Pereira, O.; Vellas, B.; Rolland, Y.; et al. Circulating levels of apelin, GDF-15 and sarcopenia: Lack of association in the MAPT study. J. Nutr. Health Aging 2022, 26, 564–570. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Terešak, P.; Lapao, A.; Subic, N.; Boya, P.; Elazar, Z.; Simonsen, A. Regulation of PRKN-independent mitophagy. Autophagy 2022, 18, 24–39. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Reiter, D.A.; Shardell, M.; Simonsick, E.M.; Studenski, S.; Spencer, R.G.; Fishbein, K.W.; Ferrucci, L. 31P magnetic resonance spectroscopy assessment of muscle bioenergetics as a predictor of gait speed in the Baltimore Longitudinal Study of Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Zane, A.C.; Reiter, D.A.; Shardell, M.; Cameron, D.; Simonsick, E.M.; Fishbein, K.W.; Studenski, S.A.; Spencer, R.G.; Ferrucci, L. Muscle strength mediates the relationship between mitochondrial energetics and walking performance. Aging Cell 2017, 16, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Mitchell, B.A.; Zampino, M.; Fishbein, K.W.; Spencer, R.G.; Ferrucci, L. Muscle mitochondrial energetics predicts mobility decline in well-functioning older adults: The Baltimore Longitudinal Study of Aging. Aging Cell 2022, 21, e13552. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Wohlgemuth, S.E.; McDermott, M.M.; Saini, S.K.; Dayanidhi, S.; Zhang, D.; Xu, S.; Kosmac, K.; Tian, L.; Ferrucci, L.; et al. Mitochondrial complex abundance, mitophagy proteins, and physical performance in people with and without peripheral artery disease. J. Am. Heart Assoc. 2023, 12, e027088. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Candia, J.; Ubaida-Mohien, C.; Lyashkov, A.; Banskota, N.; Leeuwenburgh, C.; Wohlgemuth, S.; Guralnik, J.M.; Kaileh, M.; Zhang, D.; et al. Transcriptomic and proteomic of gastrocnemius muscle in peripheral artery disease. Circ. Res. 2023, 132, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Loygorri, J.I.; Villarejo-Zori, B.; Viedma-Poyatos, Á.; Zapata-Muñoz, J.; Benítez-Fernández, R.; Frutos-Lisón, M.D.; Tomás-Barberán, F.A.; Espín, J.C.; Area-Gómez, E.; Gomez-Duran, A.; et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 2024, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.J.; Botella, J.; Genders, A.J.; Lee, M.J.C.; Saner, N.J.; Kuang, J.; Yan, X.; Granata, C. High-intensity exercise and mitochondrial biogenesis: Current controversies and future research directions. Physiology 2019, 34, 56–70. [Google Scholar] [CrossRef]
- Mesquita, P.H.C.; Lamb, D.A.; Parry, H.A.; Moore, J.H.; Smith, M.A.; Vann, C.G.; Osburn, S.C.; Fox, C.D.; Ruple, B.A.; Huggins, K.W.; et al. Acute and chronic effects of resistance training on skeletal muscle markers of mitochondrial remodeling in older adults. Physiol. Rep. 2020, 8, e14526. [Google Scholar] [CrossRef]
- Estébanez, B.; Moreira, O.C.; Almar, M.; de Paz, J.A.; Gonzalez-Gallego, J.; Cuevas, M.J. Effects of a resistance-training programme on endoplasmic reticulum unfolded protein response and mitochondrial functions in PBMCs from elderly subjects. Eur. J. Sport Sci. 2019, 19, 931–940. [Google Scholar] [CrossRef]
- Pileggi, C.A.; Hedges, C.P.; D’Souza, R.F.; Durainayagam, B.R.; Zeng, N.; Figueiredo, V.C.; Hickey, A.J.R.; Mitchell, C.J.; Cameron-Smith, D. Minimal adaptation of the molecular regulators of mitochondrial dynamics in response to unilateral limb immobilisation and retraining in middle-aged men. Eur. J. Appl. Physiol. 2023, 123, 249–260. [Google Scholar] [CrossRef]
- Tarpey, M.D.; Davy, K.P.; McMillan, R.P.; Bowser, S.M.; Halliday, T.M.; Boutagy, N.E.; Davy, B.M.; Frisard, M.I.; Hulver, M.W. Skeletal muscle autophagy and mitophagy in endurance-trained runners before and after a high-fat meal. Mol. Metab. 2017, 6, 1597–1609. [Google Scholar] [CrossRef]
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular endurance exercise promotes fission, mitophagy, and oxidative phosphorylation in human skeletal muscle independently of age. Front. Physiol. 2019, 10, 1088. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- D’Amato, M.; Morra, F.; Di Meo, I.; Tiranti, V. Mitochondrial transplantation in mitochondrial medicine: Current challenges and future perspectives. Int. J. Mol. Sci. 2023, 24, 1969. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Slavin, M.B.; Hood, D.A. Muscle mitochondrial transplantation can rescue and maintain cellular homeostasis. Am. J. Physiol. Cell Physiol. 2023, 325, C862–C884. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics consequences of mitochondrial transplantation in cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, D.; Abe, J.; Takeda, A.; Harashima, H.; Yamada, Y. Transplantation of MITO cells, mitochondria activated cardiac progenitor cells, to the ischemic myocardium of mouse enhances the therapeutic effect. Sci. Rep. 2022, 12, 4344. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Paez, H.G.; Pitzer, C.R.; Ferrandi, P.J.; Khan, M.M.; Mohamed, J.S.; Carson, J.A.; Deschenes, M.R. Mitochondria transplant therapy improves regeneration and restoration of injured skeletal muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 493–507. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzetti, E.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Picca, A. Mitochondrial Quantity and Quality in Age-Related Sarcopenia. Int. J. Mol. Sci. 2024, 25, 2052. https://doi.org/10.3390/ijms25042052
Marzetti E, Calvani R, Coelho-Júnior HJ, Landi F, Picca A. Mitochondrial Quantity and Quality in Age-Related Sarcopenia. International Journal of Molecular Sciences. 2024; 25(4):2052. https://doi.org/10.3390/ijms25042052
Chicago/Turabian StyleMarzetti, Emanuele, Riccardo Calvani, Hélio José Coelho-Júnior, Francesco Landi, and Anna Picca. 2024. "Mitochondrial Quantity and Quality in Age-Related Sarcopenia" International Journal of Molecular Sciences 25, no. 4: 2052. https://doi.org/10.3390/ijms25042052