
Multi-Omics Approaches for Freshness Estimation and Detection of Illicit Conservation Treatments in Sea Bass (Dicentrarchus Labrax): Data Fusion Applications
 , ,
, ,  , , , , , , , , and
, , , , , , , , and
Abstract
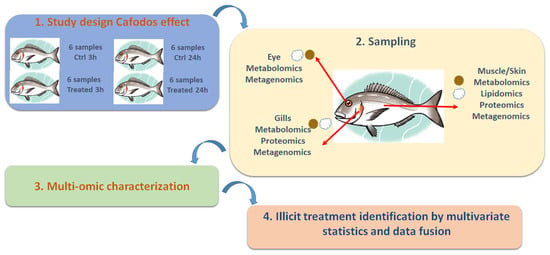
:
1. Introduction
2. Results
2.1. Multi-Omic Characterization
2.1.1. Proteomic, Lipidomic and Metabolomic Characterization
2.1.2. Metagenomics Analysis
2.2. Multivariate Statistical Analysis
2.2.1. Multiple Factor Analysis
2.2.2. BE-PLS-DA
2.3. Bioinformatics and Data Interpretation
Proteomics Ontology Data Analysis
3. Discussion
3.1. Proteomics
3.1.1. Gills
3.1.2. Skin
3.1.3. Muscle
3.2. Lipidomics
3.3. Metabolomics
3.4. Metagenomics Analysis
4. Materials and Methods
4.1. Study Design and Illicit Treatment Application
4.2. Sample Collection and Pre-Treatment
4.2.1. Proteomics
4.2.2. Lipidomics
4.2.3. Metabolomics
4.2.4. Metagenomics
4.3. Omics Determinations
4.3.1. Proteomics
4.3.2. Lipidomics
4.3.3. Metabolomics
4.3.4. Metagenomics
- -
- Controls vs. Cafodos-treated after 3 h, separately for each sampling site;
- -
- Controls vs. Cafodos-treated after 24 h, separately for each sampling site;
- -
- Controls at 3 h vs. controls at 24 h, separately for each sampling site;
- -
- Cafodos-treated at 3 h vs. Cafodos-treated after 24 h, separately for each sampling site;
- -
- All controls at 3 h vs. all Cafodos-treated samples at 3 h (all sampling sites together);
- -
- All controls at 24 h vs. all Cafodos-treated samples at 24 h (all sampling sites together).
4.4. Multivariate Data Analysis
4.5. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Purcell, S.W.; Crona, B.I.; Lalavanua, W.; Eriksson, H. Distribution of economic returns in small-scale fisheries for international markets: A value-chain analysis. Mar. Policy 2017, 86, 9–16. [Google Scholar] [CrossRef]
- Cooney, R.; de Sousa, D.B.; Fernández-Ríos, A.; Mellett, S.; Rowan, N.; Morse, A.P.; Hayes, M.; Laso, J.; Regueiro, L.; Wan, A.H.; et al. A circular economy framework for seafood waste valorisation to meet challenges and opportunities for intensive production and sustainability. J. Clean. Prod. 2023, 392, 136283. [Google Scholar] [CrossRef]
- Ye, B.; Chen, J.; Fu, L.; Wang, Y. Application of nondestructive evaluation (NDE) technologies throughout cold chain logistics of seafood: Classification, innovations and research trends. Lebensm.-Wiss. Technol. 2022, 158, 113127. [Google Scholar] [CrossRef]
- Esposito, G.; Sciuto, S.; Guglielmetti, C.; Pastorino, P.; Ingravalle, F.; Ru, G.; Bozzetta, E.M.; Acutis, P.L. Discrimination between Wild and Farmed Sea Bass by Using New Spectrometry and Spectroscopy Methods. Foods 2022, 11, 1673. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, F.; Giraudo, A.; Cavallini, N.; Esposito, G.; Merlo, G.; Geobaldo, F.; Acutis, P.L.; Pezzolato, M.; Savorani, F.; Bozzetta, E. Differentiation between fresh and thawed cephalopods using nir spectroscopy and multivariate data analysis. Foods 2021, 10, 528. [Google Scholar] [CrossRef] [PubMed]
- Currò, S.; Balzan, S.; Novelli, E.; Fasolato, L. Cuttlefish Species Authentication: Advancing Label Control through Near-Infrared Spectroscopy as Rapid, Eco-Friendly, and Robust Approach. Foods 2023, 12, 2973. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, X.; Chen, Z.; You, J.; Xiong, S. Evaluation of freshness in freshwater fish based on near infrared reflectance spectroscopy and chemometrics. Lebensm.-Wiss. Technol. 2019, 106, 145–150. [Google Scholar] [CrossRef]
- Xiang, Y.; Sun, C.; Zhao, Y.; Li, L.; Yang, X.; Wu, Y.; Chen, S.; Wei, Y.; Li, C.; Wang, Y. Label-Free Proteomic Analysis Reveals Freshness-Related Proteins in Sea Bass (Lateolabrax japonicus) Fillets Stored on Ice. Lebensm.-Wiss. Technol. 2022, 155, 112885. [Google Scholar] [CrossRef]
- Massaro, A.; Stella, R.; Negro, A.; Bragolusi, M.; Miano, B.; Arcangeli, G.; Biancotto, G.; Piro, R.; Tata, A. New Strategies for the Differentiation of Fresh and Frozen/Thawed Fish: A Rapid and Accurate Non-Targeted Method by Ambient Mass Spectrometry and Data Fusion (Part A). Food Control 2021, 130, 108364. [Google Scholar] [CrossRef]
- Stella, R.; Mastrorilli, E.; Pretto, T.; Tata, A.; Piro, R.; Arcangeli, G.; Biancotto, G. New Strategies for the Differentiation of Fresh and Frozen/Thawed Fish: Non-Targeted Metabolomics by LC-HRMS (Part B). Food Control 2022, 132, 108461. [Google Scholar] [CrossRef]
- Moser, B.; Jandrić, Z.; Troyer, C.; Priemetzhofer, L.; Domig, K.J.; Jäger, H.; Sabrina; Mayer, H.; Hann, S.; Zitek, A. Evaluation of Spectral Handheld Devices for Freshness Assessment of Carp and Trout Fillets in Relation to Standard Methods Including Non-Targeted Metabolomics. Food Control 2023, 152, 109835. [Google Scholar] [CrossRef]
- Manimaran, U.; Shakila, R.J.; Shalini, R.; Sivaraman, B.; Sumathi, G.; Selvaganapathi, R.; Jeyasekaran, G. Effect of Additives in the Shelflife Extension of Chilled and Frozen Stored Indian Octopus (Cistopus indicus). J. Food Sci. Technol. 2015, 53, 1348–1354. [Google Scholar] [CrossRef]
- Muscarella, M.; Lo Magro, S.; Campaniello, M.; Armentano, A.; Stacchini, P. Survey of Histamine Levels in Fresh Fish and Fish Products Collected in Puglia (Italy) by ELISA and HPLC with Fluorimetric Detection. Food Control 2013, 31, 211–217. [Google Scholar] [CrossRef]
- Manfredi, M.; Barberis, E.; Gosetti, F.; Conte, E.; Gatti, G.; Mattu, C.; Robotti, E.; Zilberstein, G.; Koman, I.; Zilberstein, S.; et al. Method for Noninvasive Analysis of Proteins and Small Molecules from Ancient Objects. Anal. Chem. 2017, 89, 3310–3317. [Google Scholar] [CrossRef]
- Ballabio, D.; Robotti, E.; Grisoni, F.; Quasso, F.; Bobba, M.; Vercelli, S.; Gosetti, F.; Calabrese, G.; Sangiorgi, E.; Orlandi, M.; et al. Chemical profiling and multivariate data fusion methods for the identification of the botanical origin of honey. Food Chem. 2018, 266, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Robotti, E.; Belay, M.H.; Calà, E.; Benedetto, A.; Cerruti, S.; Pezzolato, M.; Pennisi, F.; Abete, M.C.; Marengo, E.; Brizio, P. Identification of Illicit Conservation Treatments in Fresh Fish by Micro-Raman Spectroscopy and Chemometric Methods. Foods 2023, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Lin, Z.; Xu, B.; Wang, Y.; Shi, X.; Qiao, Y.; Zhang, J. Metabolomics Data Fusion between near Infrared Spectroscopy and High-Resolution Mass Spectrometry: A Synergetic Approach to Boost Performance or Induce Confusion. Talanta 2018, 189, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, A.; Pezzolato, M.; Biasibetti, E.; Bozzetta, E. Omics Applications in the Fight against Abuse of Anabolic Substances in Cattle: Challenges, Perspectives and Opportunities. Curr. Opin. Food Sci. 2021, 40, 112–120. [Google Scholar] [CrossRef]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef]
- Illikoud, N.; Gohier, R.; Werner, D.; Barrachina, C.; Roche, D.; Jaffrès, E.; Zagorec, M. Transcriptome and Volatilome Analysis during Growth of Brochothrix Thermosphacta in Food: Role of Food Substrate and Strain Specificity for the Expression of Spoilage Functions. Front. Microbiol. 2019, 10, 2527. [Google Scholar] [CrossRef]
- Yu, T.; Yi, Y.S.; Yang, Y.; Oh, J.; Jeong, D.; Cho, J.Y. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediat. Inflamm. 2012, 2012, 979105. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, X.-Y.; Li, P.; Li, Z.-C.; Zhang, C.; Sun, Y.-H.; Wang, G.-Y.; Chen, D.-D.; Lu, L.-F.; Li, S. Molecular characterization of a cyprinid fish (Ancherythroculter nigrocauda) TBK1 and its kinase activity in IFN regulation. Dev. Comp. Immunol. 2021, 114, 103805. [Google Scholar] [CrossRef]
- González-Blanco, L.; Sierra, V.; Diñeiro, Y.; Coto-Montes, A.; Oliván, M. Role of the Endoplasmic Reticulum in the Search for Early Biomarkers of Meat Quality. Meat Sci. 2023, 203, 109224. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.; Tao, W.; Zheng, S.; Wang, X.; Wang, D. Genome-wide identification, evolution and expression of the complete set of cytoplasmic ribosomal protein genes in nile tilapia. Int. J. Mol. Sci. 2020, 21, 1230. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, P.D.S.U.; Kwon, H.; Elvitigala, D.A.S.; Wan, Q.; Lee, J. Identification and characterization of cystatin B from black rockfish, Sebastes schlegelii, indicating its potent immunological importance. Fish Shellfish. Immunol. 2020, 104, 497–505. [Google Scholar] [CrossRef]
- Rajan, B.; Fernandes, J.M.; Caipang, C.M.; Kiron, V.; Rombout, J.H.; Brinchmann, M.F. Proteome Reference Map of the Skin Mucus of Atlantic Cod (Gadus morhua) Revealing Immune Competent Molecules. Fish Shellfish. Immunol. 2011, 31, 224–231. [Google Scholar] [CrossRef]
- Pinto, R.D.; Moreira, A.R.; Pereira, P.J.B.; dos Santos, N.M.S. Molecular cloning and characterization of sea bass (Dicentrarchus labrax L.) calreticulin. Fish Shellfish. Immunol. 2013, 34, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Jin, L.; Yi, X.; Dang, E.; Yang, Y.; Li, C.; Gao, T. Oxidative Stress–Induced Calreticulin Expression and Translocation: New Insights into the Destruction of Melanocytes. J. Investig. Dermatol. 2014, 134, 183–191. [Google Scholar] [CrossRef]
- Bossus, M.; Charmantier, G.; Blondeau-Bidet, E.; Valletta, B.; Boulo, V.; Lorin-Nebel, C. The ClC-3 chloride channel and osmoregulation in the European Sea Bass, Dicentrarchus labrax. J. Comp. Physiol. B 2013, 183, 641–662. [Google Scholar] [CrossRef]
- Bodinier, C.; Boulo, V.; Lorin-Nebel, C.; Charmantier, G. Influence of salinity on the localization and expression of the CFTR chloride channel in the ionocytes of Dicentrarchus labrax during ontogeny. J. Anat. 2009, 214, 318–329. [Google Scholar] [CrossRef]
- Giffard-Mena, I.; Lorin-Nebel, C.; Charmantier, G.; Castille, R.; Boulo, V. Adaptation of the sea-bass (Dicentrarchus labrax) to fresh water: Role of aquaporins and Na+/K+-ATPases. Comp. Biochem. Physiol.-A 2008, 150, 332–338. [Google Scholar] [CrossRef]
- Boutet, I.; Long Ky, C.L.; Bonhomme, F. A transcriptomic approach of salinity response in the euryhaline teleost, Dicentrarchus labrax. Gene 2006, 379, 40–50. [Google Scholar] [CrossRef]
- Wang, X.; Kültz, D. Osmolality/salinity-responsive enhancers (OSREs) control induction of osmoprotective genes in euryhaline fish. Proc. Natl. Acad. Sci. USA 2017, 114, E2729–E2738. [Google Scholar] [CrossRef] [PubMed]
- Kolbeck, S.; Ludwig, C.; Meng, C.; Hilgarth, M.; Vogel, R.F. Comparative Proteomics of Meat Spoilage Bacteria Predicts Drivers for Their Coexistence on Modified Atmosphere Packaged Meat. Front. Microbiol. 2020, 11, 209. [Google Scholar] [CrossRef]
- Han, B.; Luo, J.; Jiang, P.; Li, Y.; Wang, Q.; Bai, Y.; Chen, J.; Wang, J.; Zhang, J. Inhibition of Embryonic HSP 90 Function Promotes Variation of Cold Tolerance in Zebrafish. Front. Genet. 2020, 11, 541944. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, S.; Makridis, P.; Dimopoulos, D.; Power, D.M.; Mamuris, Z.; Moutou, K.A. Myosin light chain 2 isoforms in gilthead sea bream (Sparus aurata L.): Molecular growth markers at early stages. Aquaculture 2014, 432, 434–442. [Google Scholar] [CrossRef]
- Mato, A.; Rodríguez-Vázquez, R.; López-Pedrouso, M.; Bravo, S.; Franco, D.; Zapata, C. The First Evidence of Global Meat Phosphoproteome Changes in Response to Pre-Slaughter Stress. BMC Genom. 2019, 20, 590. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wen, H.; Qi, X.; Mao, X.; Shi, Z.; Li, J.; He, F.; Yang, W.; Zhang, X.; Li, Y. Analysis of apolipoprotein multigene family in spotted sea bass (Lateolabrax maculatus) and their expression profiles in response to Vibrio harveyi infection. Fish Shellfish. Immunol. 2019, 92, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gao, J.; Li, J.; Xue, L.; Clark, K.J.; Ekker, S.C.; Du, S.J. Functional Analysis of Slow Myosin Heavy Chain 1 and Myomesin-3 in Sarcomere Organization in Zebrafish Embryonic Slow Muscles. J. Genet. Genom. 2012, 39, 69–80. [Google Scholar] [CrossRef]
- He, Y.; Huang, H.; Li, L.; Yang, X.; Hao, S.; Chen, S.; Deng, J. The effects of modified atmosphere packaging and enzyme inhibitors on protein oxidation of tilapia muscle during iced storage. Lebensm.-Wiss.-Technol. 2018, 87, 186–193. [Google Scholar] [CrossRef]
- Kjaersgård, I.; Nørrelykke, M.; Jessen, F. Changes in cod muscle proteins during frozen storage revealed by proteome analysis and multivariate data analysis. Proteomics. 2006, 6, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, X.; Ji, M.; Wu, J.; Zhu, W.; Wang, J.; Cheng, C.; Chen, L.; Zhang, Q. Preservative effects of fish gelatin coating enriched with CUR/βCD emulsion on grass carp (Ctenopharyngodon idellus) fillets during storage at 4 °C. Food Chem. 2019, 272, 643–652. [Google Scholar] [CrossRef]
- Hwang, H.J.; Moon, C.H.; Kim, H.G.; Kim, J.Y.; Lee, J.M.; Park, J.W.; Chung, D.K. Identification and Functional Analysis of Salmon Annexin 1 Induced by a Virus Infection in a Fish Cell Line. J. Virol. 2007, 81, 13816–13824. [Google Scholar] [CrossRef]
- Dang, D.; Zhai, C.; Nair, M.N.; Thornton, K.J.; Sawalhah, M.N.; Matarneh, S.K. Tandem Mass Tag Labeling to Assess Proteome Differences between Intermediate and Very Tender Beef Steaks. J. Anim. Sci. 2022, 100, skac042. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Rubio, H.F.; Vega-López, A. Fatty acid metabolism in fish species as a biomarker for environmental monitoring. Environ. Pollut. 2016, 218, 297–312. [Google Scholar] [CrossRef]
- Alasalvar, C.; Taylor, K.D.A.; Zubcov, E.; Shahidi, F.; Alexis, M. Differentiation of cultured and wild sea bass (Dicentrarchus labrax): Total lipid content, fatty acid and trace mineral composition. Food Chem. 2002, 79, 145–150. [Google Scholar] [CrossRef]
- Reyes, M.; Rodríguez, M.; Montes, J.; Barroso, F.G.; Fabrikov, D.; Morote, E.; Sánchez-Muros, M.J. Nutritional and Growth Effect of Insect Meal Inclusion on Seabass (Dicentrarchus labrax) Feeds. Fishes 2020, 5, 16. [Google Scholar] [CrossRef]
- Montero, D.; Carvalho, M.; Terova, G.; Fontanillas, R.; Serradell, A.; Ginés, R.; Tuset, V.M.; Acosta, F.; Rimoldi, S.; Bajek, A.; et al. Nutritional Innovations in Superior European Sea Bass (Dicentrarchus labrax) Genotypes: Implications on Fish Performance and Feed Utilization. Aquaculture 2023, 572, 739486. [Google Scholar] [CrossRef]
- Syropoulou, F.; Anagnostopoulos, D.A.; Parlapani, F.F.; Karamani, E.; Stamatiou, A.; Tzokas, K.; Nychas, G.E.; Boziaris, I.S. Microbiota Succession of Whole and Filleted European Sea Bass (Dicentrarchus labrax) during Storage under Aerobic and MAP Conditions via 16S RRNA Gene High-Throughput Sequencing Approach. Microorganisms 2022, 10, 1870. [Google Scholar] [CrossRef]
- Thomas, A.; Konteles, S.J.; Ouzounis, S.; Papatheodorou, S.; Tsakni, A.; Houhoula, D.; Tsironi, T. Bacterial Community in Response to Packaging Conditions in Farmed Gilthead Seabream. Aquac. Fish. 2023, 8, 410–421. [Google Scholar] [CrossRef]
- Rosado, D.; Pérez-Losada, M.; Severino, R.; Cable, J.; Xavier, R. Characterization of the Skin and Gill Microbiomes of the Farmed Seabass (Dicentrarchus labrax) and Seabream (Sparus aurata). Aquaculture 2019, 500, 57–64. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput Microbial Community Analysis on the Illumina HiSeq and MiSeq Platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Derome, N. Evenness-Richness Scatter Plots: A Visual and Insightful Representation of Shannon Entropy Measurements for Ecological Community Analysis. mSphere 2021, 6, 10–1128. [Google Scholar] [CrossRef]
- Kim, B.R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R.E. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Gail, M.H.; Wan, Y.; Shi, J. Power of Microbiome Beta-Diversity Analyses Based on Standard Reference Samples. Am. J. Epidemiol. 2021, 190, 439–447. [Google Scholar] [CrossRef]
- Andreani, A.; Beltramo, C.; Ponzetta, M.P.; Belcari, A.; Sacchetti, P.; Acutis, P.L.; Peletto, S. Analysis of the Bacterial Communities Associated with Pupae and Winged or Wingless Adults of Lipoptena Fortisetosa Collected from Cervids in Italy. Med. Vet. Entomol. 2023, 37, 472–482. [Google Scholar] [CrossRef]
- Abascal, E.; de Rada, V.D.; Lautre, I.G.; Landaluce, M.I. Extending dual multiple factor analysis to categorical tables. J. Appl. Stat. 2013, 40, 415–428. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J.; Valentin, D. Multiple factor analysis: Principal component analysis for multitable and multiblock data sets. Wiley Interdiscip. Rev. Comput. Stat. 2013, 5, 149–179. [Google Scholar] [CrossRef]
- Benedetto, A.; Pezzolato, M.; Robotti, E.; Biasibetti, E.; Poirier, A.; Dervilly, G.; Le Bizec, B.; Marengo, E.; Bozzetta, E. Profiling of transcriptional biomarkers in FFPE liver samples: PLS-DA applications for detection of illicit administration of sex steroids and clenbuterol in veal calves. Food Control 2021, 128, 108149. [Google Scholar] [CrossRef]
- Oussama, A.; Elabadi, F.; Platikanov, S.; Kzaiber, F.; Tauler, R. Detection of olive oil adulteration using FT-IR spectroscopy nd PLS with variable importance of projection (VIP) scores. J. Am. Oil Chem. Soc. 2012, 89, 1807–1812. [Google Scholar] [CrossRef]
- Ballabio, D.; Consonni, V. Classification tools in chemistry. Part 1: Linear models. PLS-DA. Anal. Methods 2013, 5, 3790–3798. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gill | Eye | Skin | |||
|---|---|---|---|---|---|
| Family | Abundance (%) | Family | Abundance (%) | Family | Abundance (%) |
| Flavobacteriaceae | 39.00 | Pseudoalteromonadaceae | 49.00 | Moraxellaceae | 38.00 |
| Vibrionaceae | 30.00 | Comamonadaceae | 19.00 | Shewanellaceae | 21.00 |
| Pseudoalteromonadaceae | 20.00 | Shewanellaceae | 13.00 | Pseudoalteromonadaceae | 19.00 |
| Shewanellaceae | 7.00 | Moraxellaceae | 12.00 | Comamonadaceae | 13.00 |
| Moraxellaceae | 2.00 | Flavobacteriaceae | 2.00 | Flavobacteriaceae | 5.00 |
| Comamonadaceae | 2.00 | Xanthobacteraceae | 1.00 | Pseudomonadaceae | 1.00 |
| Moritellaceae | 0.23 | Propionibacteriaceae | 0.82 | Propionibacteriaceae | 1.00 |
| Rhodobacteraceae | 0.18 | Corynebacteriaceae | 0.69 | Xanthobacteraceae | 0.70 |
| Hydrogenophilaceae | 0.12 | Moritellaceae | 0.63 | Vibrionaceae | 0.22 |
| Rickettsiaceae | 0.04 | Pseudomonadaceae | 0.39 | Micrococcaceae | 0.15 |
| Tissue | Family | Max Group Mean | Log2 Fold Change | Fold Change | p-Value | FDR p-Value |
|---|---|---|---|---|---|---|
| Eye | Xanthobacteraceae | 8.1 × 102 | 6.26 | 77 | 0.0000 | 0.0005 |
| Comamonadaceae | 1.2 × 103 | 6.05 | 66 | 0.0001 | 0.0013 | |
| Oxalobacteraceae | 1.0 × 102 | 5.19 | 37 | 0.0017 | 0.0092 | |
| Corynebacteriaceae | 5.3 × 102 | 9.20 | 5.9 × 102 | 0.0041 | 0.0147 | |
| Propionibacteriaceae | 4.4 × 102 | 3.96 | 16 | 0.0042 | 0.0147 | |
| Rhizobiaceae | 1.4 × 102 | 10.43 | 1.4 × 103 | 0.0089 | 0.0234 | |
| Skin | Flavobacteriaceae | 3.5 × 102 | 6.53 | 92 | 0.0040 | 0.0228 |
| Pseudoalteromonadaceae | 4.2 × 102 | 3.22 | 9.3 | 0.0042 | 0.0228 |
| Sampling Site | Omics Sources and (Original Variables) | Omics Sources and (Selected Variables) | N° LVs | Acc% Cal | Acc% cv |
|---|---|---|---|---|---|
| Muscle 3 h | Lipid (921) + Metab (464) + Prot (66) | Lipid (45) + Metab (16) + Prot (1) | 1 | 100 | 100 |
| Lipid (921) | Lipid (18) | 1 | 100 | 100 | |
| Metab (464) | Metab (14) | 1 | 100 | 100 | |
| Prot (66) | Prot (6) | 1 | 100 | 100 | |
| Muscle 24 h | Lipid (921) + Metab (464) + Prot (66) | Lipid (11) + Metab (13) + Prot (3) | 1 | 100 | 100 |
| Lipid (921) | Lipid (27) | 1 | 100 | 100 | |
| Metab (464) | Metab (15) | 1 | 100 | 100 | |
| Prot (66) | Prot (6) | 1 | 100 | 100 | |
| Skin 3 h | Lipid (944) + Metab (384) + Prot (99) | Lipid (26) + Metab (26) + Prot (1) | 1 | 100 | 100 |
| Lipid (944) | Lipid (18) | 1 | 100 | 100 | |
| Metab (384) | Metab (24) | 1 | 100 | 100 | |
| Prot (99) | Prot (12) | 1 | 100 | 100 | |
| Skin 24 h | Lipid (944) + Metab (384) + Prot (99) | Lipid (64) + Metab (69) + Prot (14) | 1 | 100 | 100 |
| Lipid (944) | Lipid (12) | 1 | 100 | 100 | |
| Metab (384) | Metab (34) | 1 | 100 | 100 | |
| Prot (99) | Prot (4) | 1 | 100 | 100 | |
| Gills 3 h | Metab (380) + Prot (108) | Metab (59) + Prot (14) | 1 | 100 | 100 |
| Metab (380) | Metab (69) | 1 | 100 | 100 | |
| Prot (108) | Prot (12) | 1 | 100 | 100 | |
| Gills 24 h | Metab (380) + Prot (108) | Metab (7) + Prot (1) | 1 | 100 | 100 |
| Metab (380) | Metab (14) | 1 | 100 | 100 | |
| Prot (108) | Prot (14) | 1 | 100 | 100 | |
| Eye 3 h | Metab (340) | Metab (69) | 1 | 100 | 100 |
| Eye 24 h | Metab (340) | Metab (21) | 1 | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetto, A.; Robotti, E.; Belay, M.H.; Ghignone, A.; Fabbris, A.; Goggi, E.; Cerruti, S.; Manfredi, M.; Barberis, E.; Peletto, S.; et al. Multi-Omics Approaches for Freshness Estimation and Detection of Illicit Conservation Treatments in Sea Bass (Dicentrarchus Labrax): Data Fusion Applications. Int. J. Mol. Sci. 2024, 25, 1509. https://doi.org/10.3390/ijms25031509
Benedetto A, Robotti E, Belay MH, Ghignone A, Fabbris A, Goggi E, Cerruti S, Manfredi M, Barberis E, Peletto S, et al. Multi-Omics Approaches for Freshness Estimation and Detection of Illicit Conservation Treatments in Sea Bass (Dicentrarchus Labrax): Data Fusion Applications. International Journal of Molecular Sciences. 2024; 25(3):1509. https://doi.org/10.3390/ijms25031509
Chicago/Turabian StyleBenedetto, Alessandro, Elisa Robotti, Masho Hilawie Belay, Arianna Ghignone, Alessia Fabbris, Eleonora Goggi, Simone Cerruti, Marcello Manfredi, Elettra Barberis, Simone Peletto, and et al. 2024. "Multi-Omics Approaches for Freshness Estimation and Detection of Illicit Conservation Treatments in Sea Bass (Dicentrarchus Labrax): Data Fusion Applications" International Journal of Molecular Sciences 25, no. 3: 1509. https://doi.org/10.3390/ijms25031509